
Abstract
Recent advancements in sequencing and genome assembly technologies have led to rapid generation of high-quality genome assemblies for various species and breeds. Despite the importance as minipigs an animal model in biomedical research, the construction of high-quality genome assemblies of minipigs still lags behind other pig breeds. To address this problem, we constructed a high-quality chromosome-level genome assembly of the Korean minipig (KMP) utilizing multiple different types of sequencing reads and reference genomes. The KMP assembly included 19 chromosome-level sequences with a total length of 2.52 Gb and N50 of 137 Mb. Comparative analyses with the pig reference genome (Sscrofa11.1) demonstrated comparable contiguity and completeness of the KMP assembly. Additionally, genome annotation analyses identified 22,666 protein-coding genes and repetitive elements occupying 40.10% of the genome. The KMP assembly and genome annotation provide valuable resources that can contribute to various future research on minipig and other pig breeds.
Similar content being viewed by others


Background & Summary
Genome assemblies are foundational resources of various comparative genomic analyses including comparative, functional, and population analyses1. To improve the quality and accuracy of those analyses, the use of high-quality chromosome-level genome assemblies is necessary. Recently, as various types of sequencing technologies, such as long read sequencing and Hi-C sequencing, have been developed with a large amount of sequencing data accumulated, the construction of high-quality genome assemblies of various species and breeds has been accelerated2. The development of various genome assembly algorithms has also contributed to this trend3,4,5,6,7.
Since a minipig has advantageous characteristics for biomedical research, including its small body size and physiological functions similar to humans, it has become one of the most popular animal models8. In particular, the Korean minipig is the only minipig breed registered with the United Nations and Agricultural Food Organization (FAO) and utilized in many biomedical research such as xenotransplantation9. Because of the increased need to understand unique biological features of minipigs, genome assemblies of various minipig breeds, such as Göttingen minipig10 and Bama minipig11, were constructed. However, a high-quality chromosome-level genome assembly and gene annotation for the Korean minipig is still lacking.
Therefore, we constructed a chromosome-level genome assembly of the Korean minipig using various types of sequencing reads and multiple reference genomes (Fig. 1a,Table S1 and S2). A total of 10,470 contigs were generated using PacBio long reads by Canu5, of which 1,959 high-quality contigs remained after filtering and polishing steps (Table S3). To generate chromosome-level scaffolds, an improved version of RACA3 generated scaffolds from high-quality contigs using short reads, long reads, and multiple reference genomes. Existing high-quality genome assemblies of two minipigs (Bama and Göttingen), four pig breeds (Duroc, Landrace, Large white, and Meishan), cow, and goat were used as reference genomes. Scaffolding using Hi-C data was also conducted using SALSA24. The final KMP assembly was built after two polishing steps using Pilon12 (Table S3).

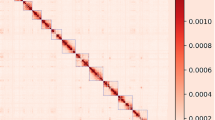
(a) Workflow for constructing the KMP assembly. (b) A Hi-C contact map of the KMP assembly (Resolution: 5 Mb; MAPQ > 30). (c) Syntenic relationships between the KMP and the pig reference genome assembly (Sscrofa11.1). Ribbons represent correspondence between chromosomes in the two genomes (Resolution: 300 Kb). (d) QV scores of 19 chromosome-level scaffolds in the KMP assembly. (e) Rates of short reads mapped to the KMP and the pig reference genome assembly (**p < 0.0002, *p < 0.02; Mann-Whitney U test).
As a result, the KMP assembly consisted of 1,042 sequences with a total length of 2.52 Gb and an N50 of 137 Mb, and 19 chromosome-level scaffolds (18 autosomes and one X chromosome) were included in the final assembly. The completeness of the KMP assembly measured by BUSCO13 using the ‘mammalia_odb9’ dataset was 93.8%. Both the contiguity and completeness of the KMP assembly were comparable to those of the pig reference genome (Sscrofa11.1; Table 1). By generating a Hi-C contact map using Juicer14, we found that the 19 chromosome-level scaffolds were clearly distinguished from each other (Fig. 1b). In addition, the quality value (QV) score for each chromosome-level scaffold of the KMP assembly was calculated using Merqury15, and the average QV score was 35.41 (Fig. 1d). Using additional short reads obtained from ten Korean minipig samples9, the mappability of the KMP assembly was confirmed, with an average of 93.63% of short reads being properly mapped (Fig. 1e,Table S4). Furthermore, the GMASS16 score between the KMP and the pig reference genome assemblies was calculated as 0.99, which confirmed the structural similarity between the two genomes. The structural similarity between these two genomes was also validated by pairwise sequence comparison and synteny analysis. All chromosome-level scaffolds in the KMP assembly except chromosome 6 formed synteny blocks with corresponding chromosomes of the pig reference genome at 300 Kb resolution without any breakpoints (Fig. 1c). Meanwhile, breakpoints detected in chromosome 6 were confirmed as real genome rearrangements in the Korean minipig (Technical Validation; Fig. 3).
To annotate genes in the KMP assembly, RNA-seq reads from 26 different tissues of the Korean minipig individual were generated (Table S1). For the annotation of protein-coding genes, we integrated RNA-seq data and gene annotation data of six species (human, mouse, pig, cow, goat, and sheep) using GeMoMa17 (Fig. 2a). As a result, a total of 22,666 protein-coding genes and 45,209 transcripts were annotated (Fig. 2b, Table 2). In addition, the average lengths of protein-coding genes, coding sequences, and protein sequences were 49,985.43 bp, 1,607.88 bp, and 534.96 bp, respectively (Table 2). To validate the quality of the gene annotation of the KMP assembly, the BUSCO score was calculated using protein sequences, and 96.4% of core mammalian genes were detected, which was comparable to the reference gene annotation (Table 2). Additionally, distributions of protein-coding genes and transcripts in the KMP assembly were similar to those in the reference genome assembly. Furthermore, the functions of 94.94% of protein-coding genes (21,519 genes) were predicted successfully using homologous gene information and the UniProtKB/Swiss-Prot database18. Four types of non-coding RNAs, including tRNA, rRNA, snRNA, and miRNA, were also identified in the KMP assembly (Fig. 2a, Table 3). Finally, repetitive elements in the KMP assembly were annotated using RepeatMasker19. As a result, 40.10% of the KMP assembly (about 1.01 Gb) was annotated as repetitive regions. Among masked repetitive elements, LINEs were the most abundant element, accounting for 23.71% of the entire genome (Fig. 2c,Table S5). The KMP assembly, which is a high-quality chromosome-level genome assembly of the Korean minipig, and its gene annotation information provide valuable resources that can contribute to various future research on minipig and other pig breeds.

(a) Workflow for annotating protein-coding and non-coding genes. (b) Genomic distributions of protein-coding genes and transcripts in chromosome-level scaffolds of the KMP and the pig reference genome assembly (Sscrofa11.1). (c) Sequence divergence of repetitive elements in the KMP assembly.
Methods
DNA and RNA sequencing
Blood sample of a male Korean minipig (27 months old) was collected with approval by the National Institute of Animal Science (NIAS) and all procedures were performed according to the ARRIVE guidelines. DNA was extracted from the collected blood sample and DNA libraries for long reads were prepared using a SMRTbell Template Prep Kit and sequenced on a PacBio Sequel system. For short read data, libraries for paired-end reads and mate-pair reads were constructed using a TruSeq Nano DNA Kit and a Nextera Mate Pair Sample Prep Kit, respectively, and sequenced on an Illumina platform. In addition, Hi-C sequencing reads were generated using the same procedure for generating paired-end reads (Table S1).
RNAs from 26 different tissues (appendix, backfat, bone marrow, brain, colon, forelimb muscle, groin, heart, hindlimb muscle, intestine, kidney, liver, lung, lymph node, nipple, pancreas, phren, pituitary gland, rib, sirloin, spinal cord, spleen, stomach, tenderloin, testis, and thymus) were also extracted using a TRIzol reagent. Sequencing libraries were then prepared using a TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina platform (Table S1).
Genome assembly
The size of the Korean minipig genome was estimated using the k-mer distribution (k = 19) calculated with Jellyfish (v2.3.0)20. Contigs were generated by connecting PacBio subreads using Canu (v1.9)5, with an estimated genome size of 2.5 G as the ‘genomeSize’ option. Only contigs supported by a minimum of 50 subreads were selected for the subsequent assembly procedure. Remaining contigs were polished using GenomicConsensus (v2.3.3; https://github.com/PacificBiosciences/GenomicConsensus) with the ‘–algorithm = arrow’ option, incorporating information from PacBio subreads mapped to contigs using pbmm2 (v1.2.1; https://github.com/PacificBiosciences/pbmm2).
To build a chromosome-level genome assembly, contigs were scaffolded using various types of sequencing data, including short reads, long reads, and Hi-C reads, as well as multiple reference genomes. Firstly, polished contigs were assembled into longer scaffolds using an improved version of RACA3 (manuscript in preparation), which integrated diverse sequencing read data and multiple reference genome information. To prepare input data for RACA, short and long read data were mapped to the polished contigs using BWA-MEM (v0.7.17-r1198)21 and pbmm2 (v1.2.1; https://github.com/PacificBiosciences/pbmm2), respectively. In addition, reference genomes of three minipig breeds (Bama, Göttingen, and Meishan), three pig breeds (Duroc, Landrace, and Large white), cow, and goat were collected from the NCBI database22 (Table S2). Using the genome assembly of Duroc (Sscrofa11.1) as a reference, pairwise whole-genome alignments were generated by LASTZ (v1.04.00)23 with ‘E = 150 H = 2000 K = 4500 L = 2200 M = 254 O = 600 Q = human_chimp.v2.q T = 2 Y = 15000’ options. Considering the divergence time against the Korean minipig, all pig breeds were used as ingroup species, while cow and goat were used as outgroup species. Secondly, scaffolds generated by RACA were further assembled using Hi-C data. For Hi-C scaffolding, Hi-C reads were aligned to scaffolds using the Arima Hi-C mapping pipeline (https://github.com/ArimaGenomics/mapping_pipeline) and SALSA24 was run with the ‘-e GATC’ option. Lastly, correction of misassemblies and the gap closing were done with short read data twice using Pilon (v1.22)12.
Genome assembly quality assessment
To assess the contiguity of the KMP assembly, assembly statistics were calculated using assembly-stats (v1.0.0; https://github.com/sanger-pathogens/assembly-stats). The completeness of genome assembly was calculated with BUSCO (v3.0.2)13 using the mammalia_odb9 dataset. Assembly statistics for the pig reference genome (Sscrofa11.1) were also calculated and benchmarked with those of the KMP assembly. To validate Hi-C mapping patterns of the KMP assembly, Hi-C reads were mapped using Juicer (v1.6)14 and the Hi-C contact map was visualized with JuiceBox (v2.3.4)24. In addition, the quality value (QV) score for each chromosome was estimated with short reads using Merqury (v1.3)15. Additional short reads from ten Korean minipig samples (five ET-type Korean minipigs and five L-type Korean minipigs) were also mapped to the KMP and pig reference genome assemblies using BWA-MEM (v.0.7.17-r1198)21. The number of mapped reads and properly mapped reads were counted using the ‘stats’ module in samtools (v1.9)25.
Next, the quality of the generated KMP assembly was validated by comparing the genomic structure between the KMP and the pig reference genome. The GMASS16 score representing structural similarity between two genome assemblies was measured using GMASS with ‘-r 100000,200000,300000,400000,500000 -s near’ options. Lastly, whole genome alignment of the KMP assembly against the pig reference genome assembly was conducted using LASTZ (v1.04.00)23 with the same options used in the ‘Genome assembly’ section. Synteny blocks were constructed at 300 Kb resolution using the synteny block detection program in InferCars26. The number of matched and mismatched bases in the syntenic regions was calculated using the Perl script (https://github.com/jkimlab/NCMD_study) provided by a previous study27.
Validation for genome rearrangement in the KMP assembly
To verify the quality of the KMP assembly, physical coverage patterns of breakpoint regions discovered through the synteny analysis were confirmed. Breakpoint regions were defined as non-syntenic regions adjacent to synteny blocks with different orders or orientations in the KMP assembly when compared to the pig reference genome. To measure base-level read coverages in breakpoint regions, short reads (paired-end and mate-pair) and long reads were mapped to the KMP assembly using BWA-MEM (v.0.7.17-r1198)21 and pbmm2 (v1.2.1; https://github.com/PacificBiosciences/pbmm2), respectively. Base-level coverage values were calculated using the ‘genomecov’ module in bedtools (v.2.28.0)28 with the ‘-bga’ option. Read coverage patterns in the breakpoint regions including the ±1~200 Kb flanking regions were visualized.
Genome annotation
For annotating protein-coding genes, RNA-seq data generated from 26 different tissues of the Korean minipig were mapped to chromosome-level scaffolds in the KMP assembly using HISAT2 (v2.2.1)29. In addition, the reference genome assembly and gene annotation data of six different species (human, mouse, pig, cow, goat, and sheep) were collected from the Ensembl database30 for homology-based gene annotation (Table S2). Using both RNA-seq and collected gene annotation data, we predicted protein-coding genes in the KMP assembly by running GeMoMa (v1.9)17 with ‘ERE.s = FR_FIRST_STRAND m = 200000 AnnotationFinalizer.r = NO GAF.f = “start = = ‘M’ and stop = = ‘*’ and (isNaN(score) or score/aa > = 4)”’ options. Subsequently, BUSCO scores were calculated for protein sequences extracted using the final KMP and the reference gene annotation by BUSCO (v3.0.2)13 with mammalian_odb9 dataset. To predict functions of protein-coding genes in the KMP gene annotation, homologous gene information identified by GeMoMa17 was used. When multiple gene functions were found for a single protein-coding gene, the function of the protein with the highest ‘pident’ value in the protein sequence alignment with vertebrate protein sequences was selected. BLASTP (v2.9.0)31 was employed for protein sequence alignment using protein sequences of vertebrate species collected from the UniProtKB/Swiss-Prot database18 (v2024_02).
For annotating non-coding genes, various types of non-coding RNAs, including tRNA, rRNA, snRNA, and miRNA, were annotated using the Rfam database32 and Infernal (v1.1.3)33 with ‘–cut_ga–rfam–nohmmonly’ options. Additionally, tRNA and rRNA were predicted with tRNAscan-SE (v2.0.5)34 and RNAmmer (v1.2)35, respectively. The final annotation was generated by merging all predictions using the Perl script (https://github.com/jkimlab/NCMD_study) provided by a previous study27.
To annotate repetitive elements, a de novo repeat library and an existing pig taxon-specific repeat library were merged as described in a previous study27. A de novo repeat library for the KMP assembly was built using RepeatModeler (v2.0.1)36, and a pig taxon-specific repeat library was extracted from the RepeatMasker (v4.0.5)19 database with the ‘queryRepeatDatabase.pl’ utility.
Data Records
The KMP assembly and gene annotation were deposited at DDBJ/ENA/GenBank under accession JBCQFQ00000000037 and FigShare38, respectively. Raw Illumina short read, PacBio long read, and Hi-C sequencing data for generating genome assembly and RNA-seq data for annotating the KMP assembly are available at NCBI SRA under accession number PRJNA110414839.
Technical Validation
To evaluate the quality of the KMP assembly, various statistics representing contiguity and completeness were measured (Table 1). The total length of the KMP assembly was 2.52 Gb, which was longer than the reference genome (2.50 Gb). The N50 of the KMP assembly was 137.31 Mb, comparable to the reference genome (138.97 Mb). A total of 19 chromosome-level scaffolds (except for the one corresponding to the Y chromosome) were constructed and Hi-C contact patterns of those scaffolds were clearly distinguished from each other. The average QV score was 35.41, with 93.8% of core mammalian genes being present in the KMP assembly (Fig. 1d,Table 1). Average rates of short reads mapped and properly mapped to the KMP assembly were 97.85% and 93.63%, respectively, higher than those of the reference genome (Fig. 1e,Table S3).
Additionally, we performed comparative analyses between the KMP assembly and the pig reference genome assembly. The GMASS score was 0.99, indicating a high similarity between the two genome assemblies. When comparing those two genome assemblies by constructing synteny blocks, most chromosome-level scaffolds in the KMP assembly showed high collinearities with corresponding chromosome assemblies in the reference genome, while several inversion events were detected in chromosome 6 (Fig. 1c). To determine whether they were caused by misassemblies or real genome rearrangement, base-level read coverages in the breakpoint regions related to the inversions were measured using short (paired-end and mate-pair) and long reads. A total of five breakpoint regions were identified and read coverage values of the breakpoint regions including the ±1~200 Kb flanking regions were visualized (Fig. 3). As shown in Fig. 3, all breakpoint regions were supported by sufficient read mapping coverages by all types of read data. In addition, physical coverages were maintained constant in boundary areas of these breakpoints. Furthermore, 99.50% of bases of the KMP assembly in the syntenic regions were matched with the pig reference assembly, while only 0.50% of bases were mismatched (Table S6).

(a) Syntenic relationship of chromosome 6 between the KMP and the pig reference genome assembly. Ribbons represent syntenic regions and colored ribbons highlight synteny blocks associated with inversion events. Arrows indicate the five breakpoint regions detected in chromosome 6. (b) Read coverage patterns of short (paired-end and mate-pair) and long reads in breakpoint regions. Colored boxes and dots represent breakpoint regions and read coverages in the boundaries of the breakpoint regions, respectively. Each breakpoint region is indicated by an arrow with the same color as in (a).
Code availability
All programs and pipelines used in this study are open-sourced. Versions and options used for the execution of individual programs are provided in the ‘Method’ section. Unless otherwise specified, default options were employed. No in-house scripts were implemented for this study.
References
Howe, K. et al. Significantly improving the quality of genome assemblies through curation. Gigascience 10, giaa153 (2021).
Chen, Q. et al. Recent advances in sequence assembly: principles and applications. Briefings in functional genomics 16, 361–378 (2017).
Kim, J. et al. Reference-assisted chromosome assembly. Proceedings of the National Academy of Sciences 110, 1785–1790 (2013).
Ghurye, J. et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS computational biology 15, e1007273 (2019).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research 27, 722–736 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18, 170–175 (2021).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome research 30, 1291–1305 (2020).
Vodička, P. et al. The miniature pig as an animal model in biomedical research. Annals of the New York Academy of Sciences 1049, 161–171 (2005).
Arora, D. et al. Multi-omics approaches for comprehensive analysis and understanding of the immune response in the miniature pig breed. Plos one 17, e0263035 (2022).
Heckel, T. et al. Functional analysis and transcriptional output of the Göttingen minipig genome. BMC genomics 16, 1–19 (2015).
Zhang, L. et al. Development and genome sequencing of a laboratory-inbred miniature pig facilitates study of human diabetic disease. iScience 19, 162–176 (2019).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963 (2014).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biology 21, 1–27 (2020).
Kwon, D., Lee, J. & Kim, J. GMASS: a novel measure for genome assembly structural similarity. BMC bioinformatics 20, 1–9 (2019).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Gene prediction: Methods and protocols, 161-177 (2019).
Boutet, E., Lieberherr, D., Tognolli, M., Schneider, M. & Bairoch, A. in Plant bioinformatics: methods and protocols 89-112 (Springer, 2007).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Marcais, G. & Kingsford, C. Jellyfish: A fast k-mer counter. Tutorialis e Manuais 1, 1038 (2012).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic acids research 50, D20 (2022).
Harris, R. S. Improved pairwise alignment of genomic DNA. (The Pennsylvania State University, 2007).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Li, H. et al. The sequence alignment/map format and SAMtools. bioinformatics 25, 2078–2079 (2009).
Ma, J. et al. Reconstructing contiguous regions of an ancestral genome. Genome research 16, 1557–1565 (2006).
Kwon, D. et al. A chromosome-level genome assembly of the Korean crossbred pig Nanchukmacdon (Sus scrofa). Scientific Data 10, 761 (2023).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature biotechnology 37, 907–915 (2019).
Martin, F. J. et al. Ensembl 2023. Nucleic acids research 51, D933–D941 (2023).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410 (1990).
Kalvari, I. et al. Non‐coding RNA analysis using the Rfam database. Current protocols in bioinformatics 62, e51 (2018).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic acids research 35, 3100–3108 (2007).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
NCBI GenBank. https://identifiers.org/ncbi/insdc.gca:GCA_039654815.1 (2024).
Wy, S. et al. KMP assembly and gene annotation. Figshare https://doi.org/10.6084/m9.figshare.25624221.v3 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP503919 (2024).
Acknowledgements
This work was supported by the Rural Development Administration of Korea (PJ01334302) and the Ministry of Science and ICT (NRF-2021M3H9A2097134 and NRF-2022R1F1A1065159).
Author information
Authors and Affiliations
Contributions
S.W.Y.: designing and performing analyses, interpreting results, and writing the manuscript. D.H.K.: designing and performing analyses, interpreting results. W.C.P.: preparing sequencing samples and generating sequencing data. H.H.C.: preparing sequencing samples and generating sequencing data. I.C.C.: preparing sequencing samples and generating sequencing data. J.B.K.: conceiving and supervising the study, designing analyses, interpreting results, and writing the manuscript. All authors: reading and approving the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wy, S., Kwon, D., Park, W. et al. A chromosome-level genome assembly of the Korean minipig (Sus scrofa). Sci Data 11, 840 (2024). https://doi.org/10.1038/s41597-024-03680-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03680-8
