
Abstract
Dasiphora fruticosa (Rosaceae), commonly known as shrubby cinquefoil, is a flowering shrub of high ornamental value yet underutilized in East Asian landscapes. Given its broad elevational distribution range, D. fruticosa serves as an ideal model for studying genetic adaptations and speciation along elevation gradients. Here, we present a high-quality chromosome-scale assembly of D. fruticosa with a genome size of 249.23 Mb and a contig N50 length of 14.01 Mb. The genome sequence contains 32,613 protein-coding genes, of which 30,643 (93.96%) were functionally annotated. Compared to the published D. fruticosa genome sequence, our assembly demonstrates higher completeness and continuity. Furthermore, comparative genomic analyses provide insights into the phylogenetic relationship and high-altitude adaptation of D. fruticosa. Overall, our study offers a valuable genetic resource for both molecular and evolutionary research on shrubby cinquefoil.
Similar content being viewed by others


Background & Summary
Rosaceae, an economically significant family of angiosperms, is renowned for its abundant edible and ornamental plants1. While high-quality genome sequences are accessible for several edible genera such as Malus, Prunus, and Fragaria, genomic resources for many ornamental plants remain limited2. Whole genome sequences represent a fundamental resource that underpins a broad range of molecular and genetic studies. Deciphering the complete DNA sequence of ornamental plants through genome sequencing facilitates the identification and characterization of genes associated with key traits relevant to landscaping, as well as the genetic basis of environmental adaptability and disease resistance3. Such insights are helpful to the molecular studies associated to the landscape applications, while enhancing our understanding of genetic diversity for conservation purposes.
Dasiphora, commonly known as shrubby cinquefoils, is a small shrub genus belonging to the tribe Potentilleae within the subfamily Rosoideae4. This genus comprises two morphologically distinct species: the yellow-petaled Dasiphora fruticosa (Fig. 1) and the white-petaled Dasiphora glabra. Both species were previously classified under Potentilla but have been recognized as an independent genus based on molecular phylogeny5,6,7,8. The presence of woody plants distinguishes Dasiphora from other genera of Potentilleae, facilitating its wide range of landscape applications.

Photographs illustrating the morphology and habitats of Dasiphora fruticosa. Photo credit: Xu Zhang and Juntong Chen.
D. fruticosa is widely distributed across cool temperate and subarctic regions, spanning altitudes from 400–5000 m in the Qinghai-Tibet Plateau (QTP)4. High-altitude environments in the QTP are characterized by freezing temperatures, high ultraviolet (UV) radiation, and hypoxia. Genome-wide studies of alpine plants revealed genomic signatures of high‐altitude adaptation often involving pathways related to abiotic stress response and biosynthesis of secondary metabolites9,10,11,12. However, due to the limited availability of alpine plant genome sequences and the restricted distribution of most species to high-altitude regions only9,10,11,12, our understanding of adaptive evolution and speciation along altitudinal gradients remains constrained. Given its extensive elevation range, D. fruticosa presents an ideal model for investigating genetic adaptations along altitudinal gradients13,14.
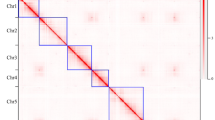
In this study, we generated a high-quality chromosome-scale genome assembly of D. fruticosa by integrating Oxford Nanopore, Illumina, and high-throughput chromosome conformation capture (Hi-C) technologies (Table 1). The genome size of D. fruticosa was estimated to be 264.18 Mb using flow cytometry (Fig. 2a,Table S1) and 264.14 Mb via 19 k-mer analysis (Fig. 2b, Table S2). The ploidy level was determined to be diploidy (Fig. 2c). We obtained 52 high-quality contigs (contig N50 = 14.01 Mb), with a total assembly size of 250.21 Mb and anchored 249.23 Mb onto seven pseudochromosomes using Hi-C data (Fig. 2d). The D. fruticosa genome contains 35.73% repetitive sequences (89.04 Mb), of which long terminal repeats (LTRs) and DNA transposons account for 13.14% and 8.51%, respectively (Table S3). We then predicted 32,613 protein-coding gene (PCG) models (Fig. 3), with functional annotation available for 30,643 PCGs (93.96%) by aligning against public protein databases (Table 1). Our genome assembly provides a valuable genomic resource for molecular studies of shrubby cinquefoils, and genetic basis of high-altitude adaptation in future studies.

Genome size estimation and chromosome assembly of Dasiphora fruticosa. (a) Flow cytometry results using Solanum lycopersicum (~900 Mb) as an internal reference for genome size estimation. (b) Distribution of k-mer depth and frequency at 19 k-mer for D. fruticosa genome. (c) Visualizations of k-mer counts output from Smudgeplot: the AB (diploid) k-mer pairs were most prominent (0.65) in the genome. (d) A Hi‐C interaction heatmap of D. fruticosa genome showing the interactions among seven pseudochromosomes.

The genomic landscape of seven pseudochromosomes of Dasiphora fruticosa. Circles from outside to inside are chromosome length (A), gene density (B), GC content (C), total LTR density (D), LTR-Gypsy density (E), LTR-Copia density (F), and syntenic blocks across chromosomes (G).
Methods
Plant material, DNA extraction and sequencing
Fresh young leaves were collected from a mature D. fruticosa plant at the Kunming Botanical Garden (Kunming, China) (102.748° E, 25.144°) and were sent to Wuhan Benagen Technology Company Limited (Wuhan, China) for genome sequencing. The voucher specimen (accession number: KUN1604678) was collected and preserved in the Herbarium of Kunming Institute of Botany (KUN). Genomic DNA was extracted using the Qiagen DNeasy Plant Mini Kit. Short-read sequencing libraries with an insert size of 500 bp were constructed using the Illumina TruSeq PCR-free HT (Illumina, San Diego, CA, USA) and were sequenced for paired-end 150 bp reads using an Illumina HiSeq. 4000 platform. For Oxford Nanopore sequencing, high-quality genomic DNA was extracted from fresh leaves using the CTAB method15. DNA quality and concentration were assessed using the NanoDrop One spectrophotometer (Thermo Fisher Scientific) and the Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). The libraries were then constructed using the SQK-LSK109 ligation kit following the standard protocol. The purified library was loaded onto primed R9.4 Spot-On Flow Cells and was sequenced using a PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK). For Hi-C sequencing, extracted DNA was first crosslinked with 40 ml of 2% formaldehyde solution to capture interacting DNA segments. Subsequently, the crosslinked DNA was digested with the DpnII restriction enzyme, and libraries were constructed and sequenced using an Illumina HiSeq. 4000 instrument with paired-end 150 bp reads.
For transcriptome sequencing, fresh tissue samples including stem, leaf, and flower were collected with three copies from the same D. fruticosa plant and immediately frozen in liquid nitrogen. Total RNA was subsequently extracted using the TRIzol® Reagent (Invitrogen, Shanghai, China). The concentration and quality of RNA were assessed using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Subsequently, paired-end cDNA libraries were prepared from mRNA enriched with anti-polyA magnetic beads, fragmented, circularized, and then subjected to PE150 sequencing on the Illumina HiSeq. 4000 platform.
Genome size estimation
The genome size D. fruticosa was estimated using two methods. First, flow cytometry was performed using Solanum lycopersicum (~900 Mb) as an internal reference, following a step-by-step protocol described by Pellicer and Leitch (2014)16. Briefly, fresh leaves of D. fruticosa and S. lycopersicum were chopped together in LB01 buffer. Nuclei were isolated, passed through a 30 mm nylon mesh, and stained with 50 mg/ml propidium iodide (PI) and 50 mg/ml ribonuclease. Samples were then analyzed on a BD FACScalibur flow cytometer, with approximately 10,000 nuclei per sample collected for three repetitions. Modifit v3.0 software was used to calculate the ratio and plotting the histogram. The genome size was estimated to be 264.18 Mb based on flow cytometry (Fig. 2a, Table S1).
Secondly, a k-mer based analysis was conducted using GCE v1.0.017. The 17 and 19 k-mer frequency-depth distribution was calculated using Jellyfish v2.2.1018 with Illumina short reads. The analysis indicated a genome size of 264.14–264.34 Mb (Fig. 2b, Table S2), consistent with the flow cytometry result. In addition, considering observed ploidy level variation in wild populations of D. fruticosa19, we estimated ploidy level of our sequenced material using Smudgeplot v0.2.520 based on the k-mer frequency output from Jellyfish. The analysis showed that AB k-mer pairs were predominant (0.65) in the genome, indicating the sequenced individual is diploid (Fig. 2c).
Genome assembly and quality control
NextDenovo v2.3.1 (https://github.com/Nextomics/NextDenovo) was employed to correct and assemble the Nanopore long reads with the following parameters: “read_type = ont, read_cutoff = 1k, genome_size = 250 m, sort_options = -m 20 g -t 15, and minimap2_options_cns = -t 8”. The initial assembly was then polished using NextPolish v1.4.121, incorporating both Illumina short reads and Nanopore long reads. After polished, the contig assembly had a total size of ~ 250.21 Mb, with a contig N50 value of 14.01 Mb (Table 1). Next, Hi-C data were used for chromosome mounting and assembly of contigs based on the 3D-DNA pipeline22 with default settings. Manual error correction was executed using Juicerbox v2.2022. A total of 249.23 Mb of the genome sequences was successfully anchored onto seven pseudochromosomes (Fig. 2d). The completeness of the assemblies was subsequently assessed by BUSCO (Benchmarking Universal Single-Copy Orthologs) assessments based on the eukaryota_odb10 database23. A total of 98.9% (94.5% single-copy BUSCOs) completeness was revealed by the analysis (Table 1). To evaluate the assembly quality, the Illumina short-reads were mapped to the assembled genome sequence using BWA v0.7.1724 with default settings, and the mapping quality score was calculated using Qualimap2 v2.2.125. A mapping rate of 94.24% and a mapping quality score of 51.15 were estimated by the analysis (Table 1).
Genome annotation
Repetitive sequences were annotated based on de novo and homology-based strategies. RepeatModeler2 v2.0.1026 was used to generate a de novo repeat library using default parameters. RepeatMasker v4.0.727 was used to run a homology search for known repeat sequences against the Repbase database. Then, RepeatMasker was employed to merge the library files from both methods and to identify the repeat contents with the parameter “-e rmblast”. The result showed that D. fruticosa genome comprises 35.73% repetitive sequences totaling 89.04 Mb, with LTRs and DNA transposons constituting 13.14% and 8.51%, respectively (Table 1 and S3).
For gene structure annotation, a combination of de novo-, homology-, and transcript-based methods were applied. RNA-seq reads were assembled using Trinity v2.1.128 with the parameter “–genome_guided_max_intron 10000”, after which coding DNA sequences (CDS) were predicted using TransDecoder v5.7.129 with default settings. De novo gene prediction was carried out with Braker2 v2.1.530. In the Braker2 run, paired-end RNA-seq reads were aligned to the D. fruticosa genome sequence using HISAT231 with default settings, and the assembled transcripts were used for training gene models in Braker2. For the homology-based method, GeMoMa v1.6.132 was employed to predict homologous sequences by mapping protein sequences of Fragaria vesca, Prunus persica, Rosa chinensis, Rubus occidentalis, Arabidopsis thaliana, and Vitis vinifera. The parameters of GeMoMa analysis were as follows: “tblastn = true, GeMoMa.Score = ReAlign, and AnnotationFinalizer.r = NO”. Finally, predicted gene models from the three methods were merged to produce consensus models using EVidenceModeler v1.1.133 following the manual provided by the authors (https://github.com/EVidenceModeler/EVidenceModeler/wiki). A total of 32,613 predicted gene modules were obtained. To validate the predicted genes, RNA-seq reads were aligned to the genome sequence of D. fruticosa using HISAT2. The reads mapped to gene regions were then quantified using featureCounts v2.0.634. The BUSCO completeness of predicted gene models was evaluated against embryophyta_odb10 datasets under the protein mode. The result showed 97.1% (92.9% single-copy BUSCOs) BUSCOs complete matched (Table 1). The Circos tool (http://www.circos.ca) was utilized to visualize gene density, GC content, and repeat content on each pseudochromosome (Fig. 3).
For the functional annotation, predicted PCGs were aligned to the UniProt and NCBI nonredundant (NR) protein databases using BLASTP (BLAST + v2.12.035) with e-value ≤ 1e-5. The functional domains of protein sequences were subsequently identified by InterProScan v5.51 based on the Pfam database36 and KEGG and GO terms of PCGs were obtained using eggNOG-mapper v2.0.137 with default settings. A total of 30,643 predicted genes (93.96%) were functionally annotated by aligning against public protein databases (Table 1).
Comparative genomic analyses
The OrthoVenn338 platform was used to identify orthologous clusters for subsequent phylogenetic and gene family contraction and expansion analyses. Nine Rosaceae species (Malus domestica, Pyrus bretschneideri, P. persica, Argentina anserina, Potentilla micrantha, F. vesca, D. fruticosa, R. chinensis and R. occidentalis), along with three outgroup species (Cannabis sativa, A. thaliana, and V. vinifera) were included. In the OrthoVenn3 analysis, highly conserved single-copy genes were aligned using MAFFT v7.52039 and trimmed by Trimal v1.240. FastTree v2.141 was used to infer the phylogenetic tree using maximum likelihood method. The SH test method was used to assess the credibility of each phylogenetic node. Divergence time was estimated based on the TimeTree5 database (http://www.timetree.org/) using MCMCTree in PAML v4.942. Subsequently, CAFÉ543 was used to estimate the expansion or contraction of gene families. Gene families with contractions and expansions were subjected to GO annotation and enrichment analysis in OrthoVenn3, matched against the UniProt database. All above analyses were implemented in the OrthoVenn3 platform and the parameters were set to defaults.
Phylogenetic results showed that Dasiphora exhibits a closer relationship to Fragaria than to Potentilla, with an estimated divergence time of approximately 15.8 million years ago (Fig. 4). Subsequently, our analysis identified 202 gene families that had contracted and 120 that had expanded (Fig. 4). Gene Ontology (GO) enrichment analyses revealed that these expanded gene families are primarily associated with response to oxidative stress, photosynthesis light reaction, response to UV-B, response to cold, and protein phosphorylation (Table S4). These pathways are primarily associated with abiotic stress response and likely play crucial roles in the adaptation of D. fruticosa to extremely cold, high UV radiation, and hypoxic environments of high altitudes.

Phylogenomic results among nine Rosaceae species. Gene family expansion (orange) and contraction (blue) information is labeled on the tree.
Data Records
Oxford Nanopore, Hi-C sequencing, Illumina, and RNA-seq data generated in this study have been deposited at the NCBI Sequence Read Archive (SRA) under Bioproject PRJNA113109844 and the China National GeneBank Database (CNGBd, https://db.cngb.org/) with accession number CNX0946916-CNX094691945,46,47,48. The genome assembly has been deposited at GenBank under the accession JBEWQC00000000049. The annotation files of the genome are available at the Figshare database: https://doi.org/10.6084/m9.figshare.25272223.v150.
Technical Validation
Genome assembly validation
The quality of the D. fruticosa genome assembly was evaluated using three methods. First, both flow cytometry and a k-mer based estimation of genome size were very similar. A final genome assembly size also revealed a close match to the estimated genome size. Secondly, the BUSCO assessment of the genome assembly indicated a high level of completeness, with 98.9% (94.5% single-copy BUSCOs) complete matches to the embryophyta_odb10 dataset. Finally, Illumina data mapping was employed to assess assembly quality. The results showed a mapping rate of 94.24% and a mapping quality score of 51.15 (corresponding to a base accuracy of 99.999%), suggesting that the genome is of high quality.
Genome annotation validation
Genome annotation was conducted using transcript-based, de novo, and homology-based prediction methods. These methods led to the prediction of 32,613 gene models. RNA-seq read mapping revealed that 29,280 genes (95.55% of all predicted genes) were detected with mapped reads in at least one of the three tissues. Functional annotation was available for 30,643 genes (93.96%) through alignment with public protein databases. The BUSCO analysis showed that the annotated protein coding genes of the D. fruticosa genome had a complete match rate of 97.1% (92.9% single-copy BUSCOs) with the embryophyta_odb10 dataset. In addition, our phylogenetic result based on PCGs was consistent with previous studies supporting a closer relationship between Dasiphora and Fragaria than with Potentilla5,6,7,8. Functional analysis of expanded gene families revealed genomic signatures of high-altitude adaptation involving pathways related abiotic stress response, in line with previous findings9,10,11,12.
Comparison with a published D. fruticosa genome
We further compared the statistics of the D. fruticosa genome assembly and annotation from our present study with those of a recent study14 (Table 1). Overall, both assemblies demonstrated similar quality, with our genome assembly showing higher completeness and continuity. Specifically, our genome assembly exhibited longer contig length and anchored genome length, along with higher BUSCO completeness compared to the published D. fruticosa genome. Moreover, our study provided flow cytometry data to validate the genome size and estimates ploidy level of our sequenced material. These results are crucial for ensuring the reliability of our genome assembly, particularly in light of observed ploidy level variation in wild populations of D. fruticosa19.
Code availability
No custom code was used for this study. All data analyses were executed utilizing publicly available bioinformatics software, with the specific version detailed in the Methods section. Except for the parameter settings specified in the Methods section, all software parameters were set to default.
References
Hummer, K. E. & Janick, J. in Genetics and Genomics of Rosaceae (eds Kevin M. Folta & Susan. E. Gardiner) 1–17 (Springer New York, 2009).
Li, M., Xiao, Y., Mount, S. & Liu, Z. An Atlas of Genomic Resources for Studying Rosaceae Fruits and Ornamentals. Frontiers in Plant Science 12 (2021).
Zheng, T., Li, P., Li, L. & Zhang, Q. Research advances in and prospects of ornamental plant genomics. Horticulture Research 8, 65 (2021).
Li, C., Hiroshi, I. & Ohba, H. in Flora of China Vol. 9 (eds C.Y. Wu, P.H. Raven, & D.Y. Hong) Potentilla Linnaeus (Beijing, China: Science Press & St. Louis USA: Missouri Botanical Garden, 2003).
Davidson, C. G. & Lenz, L. M. Experimental taxonomy of Potentilla fruticosa. Can J Bot 67, 3520–3528 (1989).
Dobeš, C. & Paule, J. A comprehensive chloroplast DNA-based phylogeny of the genus Potentilla (Rosaceae): Implications for its geographic origin, phylogeography and generic circumscription. Molecular Phylogenetics and Evolution 56, 156–175 (2010).
Chen, X. et al. Molecular systematics of Rosoideae (Rosaceae). Plant Systematics and Evolution 306, 9 (2020).
Li, Q.-Q., Khasbagan, Zhang, Z.-P., Wen, J. & Yu, Y. Plastid phylogenomics of the tribe potentilleae (Rosaceae). Molecular Phylogenetics and Evolution 190, 107961 (2024).
Birkeland, S., Gustafsson, A. L. S., Brysting, A. K., Brochmann, C. & Nowak, M. D. Multiple Genetic Trajectories to Extreme Abiotic Stress Adaptation in Arctic Brassicaceae. Mol Biol Evol 37, 2052–2068 (2020).
Chen, J. H. et al. Genome-wide analysis of Cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat Commun 10, 5230 (2019).
Zhang, T. et al. Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc Natl Acad Sci USA 116, 7137–7146 (2019).
Zhang, X. et al. Genomic convergence underlying high-altitude adaptation in alpine plants. J Integr Plant Biol 65, 1620–1635 (2023).
Ma, Y.-Z. et al. Phylogeography of the genus Dasiphora (Rosaceae) in the Qinghai-Tibetan Plateau: divergence blurred by expansion. Biological Journal of the Linnean Society 111, 777–788 (2014).
Yang, F.-S. et al. Signatures of Adaptation and Purifying Selection in Highland Populations of Dasiphora fruticosa. Molecular Biology and Evolution 41, msae099 (2024).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical bulletin (1987).
Pellicer, J. & Leitch, I. J. in Molecular Plant Taxonomy: Methods and Protocols (ed Pascale Besse) 279-307 (Humana Press, 2014).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv:1308.2012 (2013).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Elkington, T. T. Cytotaxonomic variation in potentilla fruticosa L. New Phytologist 68, 151–160 (1969).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432 (2020).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, Unit 4 10 (2009).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. https://github.com/TransDecoder/TransDecoder.
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3, lqaa108 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res 44, e89 (2016).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 38, 5825–5829 (2021).
Sun, J. et al. OrthoVenn3: an integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Research 51, W397–W403 (2023).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 5, e9490 (2010).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591 (2007).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP517649 (2024).
China National GeneBank Database (CNGBd) https://db.cngb.org/search/experiment/CNX0946916/ (2024).
China National GeneBank Database (CNGBd) https://db.cngb.org/search/experiment/CNX0946917/ (2024).
China National GeneBank Database (CNGBd) https://db.cngb.org/search/experiment/CNX0946918/ (2024).
China National GeneBank Database (CNGBd) https://db.cngb.org/search/experiment/CNX0946919/ (2024).
Zhang, X. Dasiphora fruticosa isolate XZ-2024, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBEWQC000000000 (2024).
Zhang, X. A high-quality genome of the shrubby cinquefoil (Dasiphora fruticosa). figshare https://doi.org/10.6084/m9.figshare.25272223.v1 (2024).
Acknowledgements
This work was supported by grants from the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502), the Key Research and Development Program of Yunnan Province (202403AC100028), the Young Scholars Program of Wuhan Botanical Garden (E3551G01) and the CAS “Light of West China” Program (xbzg-zdsys-202319).
Author information
Authors and Affiliations
Contributions
H.W., H.S. and X.Z. developed the idea and designed the experiment; J.C., B.R., and T.D. collected the plant materials; X.Z., J.C., H.G., S.G., Y.S. and T.D. performed the statistical analyses; X.Z. interpreted the results and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Chen, J., Guo, H. et al. A high-quality genome assembly of the shrubby cinquefoil (Dasiphora fruticosa). Sci Data 11, 950 (2024). https://doi.org/10.1038/s41597-024-03781-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03781-4
