
Abstract
Tritrichomonas foetus is a parasitic protist responsible for bovine trichomonosis, a reproductive disease associated with significant economic burden to the livestock industry throughout the world. Here, we present a chromosome-level reference genome of T. foetus -KV-1 (ATCC 30924) using short-read (Illumina Miseq), long-read (Oxford Nanopore) and chromatin-linked (Hi-C) sequencing. This is the first chromosome-level genome of a parasitic protist of the order Tritrichomonadida and the second within the Parabasalia lineage, after Trichomonas vaginalis, the human-associated causative agent of the sexually transmitted infection in humans. Our constructed genome is 148 Mb in size, with a N50 length of the scaffolds of 22.9 Mb. The contigs are anchored in five super-scaffolds, corresponding to the expected five chromosomes of the species and covering 78% of the genome assembly. We predict 41,341 protein-coding genes, of which 95.10% have been functionally annotated. This high-quality genome assembly serves as a valuable reference genome for T. foetus to support future studies in functional genomics, genetic conservation and taxonomy.
Similar content being viewed by others

Background & Summary
Bovine trichomonosis is a worldwide occurring, to the WOAH/OIE notifiable, venereal disease of cattle. The causative agent is the flagellate-like parasite Tritrichomonas (T.) foetus1. This parasite exists exclusively in the trophozoite stage and reproduces by binary longitudinal fission, without sexual reproduction. It colonises the epithelial surface of the lumen and crypts of the prepuce and penis and is transmitted during mating by asymptomatic bulls2. Infected cows and heifers, in contrast, present with premature embryonic death, uterine discharge, pyometra, irregular oestrus cycles and infertility3,4. The resulting reproductive failure not only drastically reduces the breeding efficiency in dairy and beef cattle2, it is also accompanied by significant financial damage due to reduced milk and calf production. According to estimates the losses caused by T. foetus may amount to around 1 billion US dollars per year in the US alone5,6.
T. foetus has been observed in other animal species such as domestic cats, horses and roe deer as well as swine; goats, dogs, rabbits and guinea pigs can be experimentally infected7. Human T. foetus infections causing meningoencephalitis and peritonitis in immunocompromised and immunosuppressed individuals have also been reported8. To date, the taxonomy of trichomonad parasites including the avian pathogen Trichomonas (T.) gallinae, the human parasitic pathogens Pentatrichomonas hominis, T. tenax and T. vaginalis has not been extensively studied. It is still a matter of debate to what is a true species as observed for T. suis, a commensal of pigs, and bovine T. foetus. Both parasites are indistinguishable morphologically, serologically and antigenically7. No chromosome-level genome for T. foetus exists so far and the two publicly available genome assemblies of T. foetus are too highly fragmented to obtain a descriptive reference genome needed for genetic studies, genomics and taxonomy9,10.
The present study describes the application of Illumina short-read, nanopore long-read and Hi-C sequencing methods to construct the genome of T. foetus KV-1 (ATCC 30924) at the chromosome-level. The T. foetus KV-1 genome is 148 Mb-long with an N50 scaffold length of 22.9 Mb, with 115 Mb of the assembled genome sequences mapped to five chromosomes. A total of 41,341 protein-coding genes were discovered. We incorporated the analysis steps described below into a fully automated pipeline for the characterisation of T. foetus genomics. This open-source pipeline is available at https://gitlab.com/FLI_Bioinfo/tricho-workflow and should enable laboratories not only to replicate our analytical approach, but also to apply this approach to other T. foetus strains. Although we present a high-quality genome at the chromosome scale, we anticipate that ongoing advancements in sequencing technologies, as well as assembly and annotation algorithms, will continue to enhance the precision and completeness of this genomic dataset.
Methods
T. foetus KV-1 sequencing and data pre-processing
Genomic DNA was obtained from T. foetus KV-1 cultivated in InPouch® TF-Bovine (Megacor; Austria) to a concentration of 1.5 *106/mL using the UNSET lysis buffer and subsequent phenol: chloroform extraction method as described in11. The DNA quality was assessed using a NanoDrop Spectrophotometer (Thermo Fisher, USA), and quantified by a Qubit Fluorometer (Invitrogen, USA). For long-read sequencing, we used the R9.4.1 sequencing chemistry from Oxford Nanopore Technologies (ONT) on a GridIon platform with the ligation kit SQK-LSK109. The ONT data were collected and base-called with Guppy 6.2.11 using the sup-accurate model. This resulted in a cumulative data set of 1.3 million reads with a total length of 9.40 billion bases and an average length of 7.05 kb. Subsequent trimming and filtering of the ONT data using Porechop12 (default mode) and filtlong13 (-min_length 1000), respectively, yielded 826,311 reads with an average length of 11 kb and cumulative base count of 9 billion bases (Table 1). Illumina sequencing was done on a MiSeq machine (Illumina, USA) using the XT DNA Library kit, generating paired-end short reads of 300 bp. After Illumina sequencing, 52 million reads with a cumulative base count of 12.3 billion bases of raw sequences were obtained for T. foetus KV-1. The accuracy of the Illumina reads was improved by performing adapter trimming and quality control with fastp (v0.23.2)14. This left a total of 51.4 million reads with a total number of 11.9 billion bases and a Q30 of 88.3%.
De novo genome assembly, Hi-C scaffolding and quality assessment
The de novo genome assembly was based on long-read assembly followed by polishing with short reads to ensure the contiguity and accuracy of the genome. The long-read assembly was based on nanopore reads with a length of at least 1000 bases. We then evaluated the performance of the four different assemblers on our data: flye15 (v 2.9), wtdbg216 (v2.5), Raven17 (v1.8.3) and Shasta18 (v0.11.1) (Table S1). The predicted genome size of the respective assemblers varied between 109 and 220 Mb, as did the contiguity of the genomes in terms of number of contigs (range 207 to 5992), contig N50 (range 68 Kb to 2.6 Mb) and maximum contig size (range 916 Kb to 10,7 Mb) (Table S1). The assembly results of the two assemblers Wtdbg2 (producing the largest genome with high fragmentation; Table S1) and flye (smallest genome with improved contiguity; Table S1) were merged using quickmerge19 (v0.3) based on a sequence overlap of >5 kb between assemblies. The flye assembly was then used as a query and the wtdbg2 assembly as a reference in order to create larger, contiguous genome segments. Multiple rounds of assembly polishing were subsequently performed, first with long-read using two iterations of racon20 (v1.5.0; with minimap221 v2.24), followed by two rounds of short-read polishing with the fastp-driven Illumina reads using Polypolish22 (v0.50; with bwa v0.7.17). A total of 339,488 substitution errors in the initial assembly were corrected with Illumina data resulting in a final consensus quality of 99.9%. We estimated the average read depth per contig by mapping the nanopore reads to the polished contigs using minimap221 (v2.24), followed by qualimap23 (v 2.2.2a) to evaluate the mapping quality. Finally, we discarded the contigs with a coverage depth of less than 7-fold. This approach resulted in a total number of 670 contigs (total size 148,7 Mb) which were then selected for scaffolding.
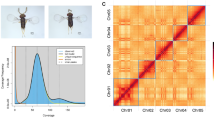
To create a chromosome level assembly, Hi-C proximity ligation data were used to anchor, order and orient the assembled contigs. In-situ Hi-C sequencing was accomplished with the EpiTect Hi-C Kit (Qiagen). In brief, two in-situ Hi-C libraries were prepared, including steps for cell crosslinking, cell lysis, chromatin digestion, biotin labelling, proximal chromatin DNA ligation and DNA purification. Hi-C sequencing using the Illumina MiSeq machine generated a total of 49.6 million raw reads with a base count of 14.9 billion bases. After data filtration with fastp (v0.23.2), a total of 45.7 million clean reads with a base count of 12.3 billion bases were retained. The ARIMA Genomics Hi-C Mapping Pipeline (https://github.com/ArimaGenomics/mapping_pipeline; accessed February 2024) was then applied to identify pairs of reads originating from physically interacting genomic regions. This pipeline aligns Hi-C reads with bwa-mem24 (v0.7.17) in single-end read mode, followed by trimming the 3′ end of reads marked as chimeric or spanning ligation junctions. Paired reads were filtered based on the mapping quality using samtools25 (v1.19.2) and PCR duplicates were eliminated with Picard26 (v 3.5.3). As no reference was available, YAHS27 (Yet another Hi-C scaffolding tool, v1.2a.2) was used to assemble the draft genome into a chromosome candidate utilising aligned Hi-C data in BAM format. Visualisation of the assembler’s Hi-C maps was obtained using HapHic plot28 (v1.0.3) (Fig. 1).

Results of Hi-C Scaffolding of T. foetus KV-1 genome assembly. (A) Genome-wide and (B) separate plots of the Hi-C interaction mapping of the five chromosome-level scaffolds in the T. foetus KV-1 Hi-C assembly. (C,D) Quality control results showing the contiguity parameters of the Hi-C scaffold and contigs of the T. foetus KV-1 genome generated in the study, compared to other available genomes of T. foetus at NCBI. The results indicate improved contiguity of our genome assembly.
The final genome assembly of T. foetus KV-1 comprised 148,7 Mb, with Hi-C analysis revealing five super-scaffolds (sizes 12, 20, 22, 26 and 33 Mb) corresponding to the expected five chromosomes of T. foetus29,30 (Fig. 1). The scaffolds anchored 78% (115 Mb) of the genome assembly, with a total N50 of 22.9 Mb and 2.18 Mb for scaffold and contig lengths, respectively. The average GC content of the genome assembly was 30.75% (Table 2, Fig. 2).

Circos plot depicting the genomic features of T. foetus KV-1. The five large scaffolds of the genome are shown on an MB scale (circle 1) followed by representation of the predicted annotated coding genes in clockwise (circle 2) and anticlockwise (circle 3) orientations. Circles 4 and 5 show the coverage pattern of short-reads and long reads across the genome, respectively. Genomic areas with abnormal coverage are highlighted with blue or red squares, denoting for excessively high (>150-fold) or low (<15-fold) coverage, respectively. Circle 5 shows repeat regions in-between the five scaffolds that are more than 20 kb in size.
This final version of the genome assembly of T. foetus KV-1 showed a considerable improvement over other publicly available T. foetus genomes based only on Illumina9 (strain K; accession, GCA_001839685.131) or PacBio data10 (strain Belfast; accession, GCA_905133005.132) (Table S2). Our assembly showed high contiguity with an improved scaffold N50 of 22 Mb. Table S2 shows genome statistics of the assembled genome along with the other publicly-available Parabasalia genomes at NCBI. The T. foetus KV-1 genome assembled in the present study is comparable in size with the T. foetus Belfast genome (146 Mb)15. The estimated genome size of T. foetus KV-1 is smaller than the genome size of T. vaginalis (160–180 Mb)33. We confirmed the smaller genome size for T. foetus KV-1 by flow cytometry (Supplementary Fig. S1). We also confirmed the absence of bacterial or archaeal contamination in our assembly (Supplementary Fig. S2). Finally, we compared our genome to all other genomes of the order Parabasalia using a set of 255 marker genes (Benchmarking Universal Single-Copy Orthologs; BUSCOs34 v5.5.0) from the eukaryota_odb10 dataset to assess the completeness results of our genome. A completeness score of 52.6% (138 complete BUSCOs) was obtained. This completeness value for T. foetus KV-1 is in a similar range as all the other Parabasalia genomes (Table S2; Fig. 3), suggesting that some integrated eukaryotic BUSCO markers may be missing in Parabasalia or that the gene prediction method applied by BUSCO is currently insufficient for these genomes. The recently described tool OMARK (v0.3.0) with the Eukaryota ancestral clade revealed a completeness of 66.3% (660 of the 995 conserved Hierarchical Orthologous Groups, HOGs). The analysis showed that 26.13% of these HOGs were single-copy, 40.2% were duplicated, and 33.67% were missing. Whole proteome analysis revealed that 14,343 out of 41,341 proteins (34.69%) showed consistent lineage placement.

Comparison of the BUSCO assessment of genome completeness for the T. foetus KV-1 genome assembled in this study in comparison to all other publicly available genomes of Parabasalia using the eukaryote_odb10 data set.
K-mer-based approximation of genome size
The k-mer-based approximation (kmer length between 11 and 33-mers) of the genome size of T. foetus KV-1 using Illumina data resulted in a genome size of ~58 Mb. This approximation employed Jellyfish35 (v2.3.09) and GenomeScope36 (v1.0.08), with an estimated heterozygosity rate of ~1.86% (Supplementary Fig. S3). This underestimation of the genome size might be caused by the complexity of the genome, non-uniform coverage of the genome structure, or an excessive abundance of highly repetitive regions.
Repeat annotation
Repetitive sequences were identified through a combination of de novo and structure-based predictions. We predicted tandem repeats with the Tandem Repeats Finder tool37 (v4.09) (Table 3). In addition, we used RepeatModeler238 (v2.0.5) with the underlying tools, RECON (v1.08), RepeatScout (v 1.0.6), LTR_retriever (v2.9.0), and LTRharvest (genometools v1.6.4), to construct de novo species-specific transposable elements (TE) libraries for repeat annotation. This TE library was subsequently employed by RepeatMasker39 (v4.1.6; https://www.repeatmasker.org/) for the identification of both known and novel TEs in the T. foetus KV-1 genome. The T. foetus KV-1 genome was highly enriched for repetitive elements with a total of 60.84 Mb identified as repeat elements representing 48.93% of the full genome. These repeat elements were distributed unevenly along the scaffolded chromosomes. Of the repetitive elements, DNA transposons were the most common and accounted for ~30% of the genome. Other categories of repetitive sequences included retroelements (2.85%), LTR elements (2.28%), LINEs (0.56%) and simple repeats (0.95%). A high proportion (16.19%) of identified repeat elements were unclassified which may be accounted by the lack of studies on the repeats in Parabasalia. Additionally, minor proportions of repetitive elements were attributed to rolling-circles and SINEs. Finally, the total number and length of microsatellites predicted in T. foetus KV-1 with Krait40 (v1.5.1) were 69,977 and 5.393,631 bp, respectively.
Annotation of non-coding RNA genes
Infernal41 (v1.1.5) was used to search for homologues of structural RNAs through alignment with the Rfam RNA database42 (v14.9) (http://rfam.xfam.org/). In this release of the Rfam database, 4108 RNA families represented by covariance models (CMs) are available. Searching the Rfam CMs with cmscan41 (v1.1.5) identified 517 high-scoring structural RNAs hits in the T. foetus KV-1 genome including 377 transfer RNAs (tRNAs) and 133 ribosomal RNAs (rRNAs) (params:–cut_ga–nohmmonly; removing the hits with high-scoring-overlaps; Table 2, Table S3).
Gene prediction
The de novo prediction of the protein-coding genes was done using BRAKER43,44,45,46,47,48,49,50,51,52,53 (v3.0.8) based on the repeats-soft-masked genome from RepeatMasker and the OrthoDB54 database of protein sequences. BRAKER applies GeneMark-ES (v4.72), with long genes predicted by GeneMark-ES serving as input for training AUGUSTUS (v3.5.0)44. Then, ab initio prediction of gene structures is performed with AUGUSTUS44. Table 3 shows the annotation features of the T. foetus KV-1 genome. In total, 41,341 protein-coding genes were identified, including 36,149 monoexonic and 3,035 multi-exonic genes. A maximum of two exons was identified per gene. The average gene length was 1490 bp (range 164 to 29,276 bp). The average exon length was 1349 bp (range 2 to 14942 bp). The average intron length was 175 bp (range 5–898 bp). Figure 2 shows the density of predicted genes, repetitive sequences, and alignment rates of the sequencing reads across the five chromosomes of the T. foetus KV-1 genome. Together, these results offer a foundation for further analysis of the genetic architecture of T. foetus.
Functional genome annotation
Functional genome annotation was performed by comparing the protein sequences of the predicted genes with the functional databases NR protein database, SWISS-PROT55, Gene Ontology (GO)56, eggNOG57 and KEGG58 using DIAMOND Blastp52 (e-value 1.0e-3) through the Functional Analysis Module of OmicsBox59 (v3.0.30). Additionally, InterProScan60 (v5.69–101.0) was used with the EMBL-EBI version of InterPro61 for the functional analysis of proteins (Table S4). Of the 41,341 predicted protein sequences, 37,751 had BLAST hits in the NR database, 31,954 in the RefSeq database and 19,555 in the SWISS-PROT database. 41,339 predicted protein sequences were identified with InterProScan. 24,714 (59.7%) predicted protein sequences were mapped with Gene Ontology Terms, of these, 23,494 (56.8%) sequences with Enzyme Code Annotations. 26,868 (53%) of the predicted sequences generated eggNOG results and could be assigned to OG functional Categories. The KEGG database identified 411 pathways, with 11,905 protein sequences linked to these pathways, while the reactome database62 identified 2480 pathways including 2223 sequences. The GO classification of the functionally annotated genes within three main GO domains showed that 49.5% (n = 21,066) of the genes belong to “biological process”, 51% (n = 21,680) to “molecular function”, and 61,9% (n = 26,318) to “cellular component” (Fig. 4). The most prominent GO terms at level 2 for biological process classes were “cellular process” (39%, n = 16,064) and “metabolic process” (31%, n = 12847). Similarly, for the cellular component domain, the main GO classes identified were “cellular anatomical entity” (49%, n = 20 134) and “protein-containing complex” (14%, n = 5786). For molecular function GO terms at level 2, “binding” (36%, n = 14,719) and “catalytic activity” (25%, n = 10,201) were the primary classes involved (Table S5).

Bar plots depicting the Gene Ontology (GO) classification of functionally annotated genes in the Hi-C assembly. The distributions of GO terms are presented within the three main GO domains including “biological process” (green), “molecular function” (blue), and “cellular component” (yellow).
Data reproducibility through automated workflow
In addition to the T. foetus KV-1 strain sequenced in this study, it is to be expected that more high-quality genome sequences will become available for this species. The availability of multiple genome sets will considerably increase the knowledge on genetic diversity within T. foetus strains of different geographic origin and animal hosts. To ensure data reproducibility and facilitate future genome studies, we have developed an automated, open-source, Snakemake63 pipeline for the analysis of T. foetus genomes, available at https://gitlab.com/FLI_Bioinfo/tricho-workflow. This pipeline incorporates the assembly and annotation steps used in this work.
Data Records
All raw data of the whole genome of T. foetus KV-1 have been deposited at the National Center for Biotechnology Information (NCBI) under the BioProject accession number PRJNA112362664; BioSample accession number SAMN41816898. The short and long read sequences have been deposited in the SRA accessions SRR29430452 and SRR29430454 respectively. The Hi-C sequencing data have been deposited in the SRA accession SRR29430453. The final genome assembly has been deposited in GenBank at JBEJVY000000000.
Technical Validation
To validate the genome assembly, QUAST65 (v5.0.2) was utilised to report assembly contiguity metrics, followed by BUSCO34 (v5.1.2), compleasm66 (v0.2.6) and OMArk67 (v0.3.0) to assess the completeness of the final scaffolded genomes. Additionally, we calculated the percentage of short reads mapping to the final scaffolded assembly. For this, Illumina data were trimmed with fastp14 and mapped using bwa mem24, and mapping quality was assessed with Qualimap23. Of the Illumina QC-passed reads, 99.98% (n = 24,967,332) were primarily mapped, with 99.65% (n = 24,831,090) properly paired. Similarly, long-read sequencing data were mapped to the genome, resulting in a mapping rate of 99.90%. Finally, the genome assembly with SPAdes68, that was based solely on Illumina reads, was fully contained within the final assembly from the long-read data. No evidence of contamination with foreign DNA from a different taxon was detected in the assembly based on metagenomics binning using MetaBAT269.
Code availability
All software and tools in this study were used with their default parameters, unless otherwise detailed. The workflow used for the assembly and annotation is available at https://gitlab.com/FLI_Bioinfo/tricho-workflow.
References
Michi, A. N., Favetto, P. H., Kastelic, J. & Cobo, E. R. A review of sexually transmitted bovine trichomoniasis and campylobacteriosis affecting cattle reproductive health. Theriogenology 85, 781–791, https://doi.org/10.1016/j.theriogenology.2015.10.037 (2016).
Cobo, E. R., Corbeil, L. B. & BonDurant, R. H. Immunity to infections in the lower genital tract of bulls. Journal of Reproductive Immunology 89, 55–61, https://doi.org/10.1016/j.jri.2011.02.002 (2011).
Skirrow, S. Z. & Bondurant, R. H. Treatment of bovine trichomoniasis with ipronidazole. Aust Vet J 65, 156, https://doi.org/10.1111/j.1751-0813.1988.tb14446.x (1988).
BonDurant, R. H. Pathogenesis, Diagnosis, and Management of Trichomoniasis in Cattle. Veterinary Clinics of North America: Food Animal Practice 13, 345–361, https://doi.org/10.1016/S0749-0720(15)30346-7 (1997).
Martin, K. A., Henderson, J. & Brewer, M. T. Bovine Trichomonosis Cases in the United States 2015-2019. Front Vet Sci 8, 692199, https://doi.org/10.3389/fvets.2021.692199 (2021).
Gifford, C. A. et al. Factors important for bull purchasing decisions and management in extensive rangeland production systems of New Mexico: a producer survey. Translational Animal Science 7, https://doi.org/10.1093/tas/txac167 (2022).
Slapeta, J. et al. Comparative analysis of Tritrichomonas foetus (Riedmüller, 1928) cat genotype, T. foetus (Riedmüller, 1928) cattle genotype and Tritrichomonas suis (Davaine, 1875) at 10 DNA loci. Int J Parasitol 42, 1143–1149, https://doi.org/10.1016/j.ijpara.2012.10.004 (2012).
Yao, C. Diagnosis of Tritrichomonas foetus-infected bulls, an ultimate approach to eradicate bovine trichomoniasis in US cattle? Journal of medical microbiology 62, 1–9, https://doi.org/10.1099/jmm.0.047365-0 (2013).
Benchimol, M. et al. Draft Genome Sequence of Tritrichomonas foetus Strain K. Genome announcements 5, https://doi.org/10.1128/genomeA.00195-17 (2017).
Senior, E. M. A reverse vaccinology approach to identifying vaccine candidate antigens for bovine Trichomoniasis. (The University of Liverpool (United Kingdom), 2020).
Horner, D. S., Hirt, R. P., Kilvington, S., Lloyd, D. & Embley, T. M. Molecular data suggest an early acquisition of the mitochondrion endosymbiont. Proc Biol Sci 263, 1053–1059, https://doi.org/10.1098/rspb.1996.0155 (1996).
Wick, R. Porechop: Adapter trimmer for Oxford Nanopore reads. Github https://github.com/rrwick (2017).
Wick, R. Filtlong: Quality filtering tool for long reads. GitHub https://github.com/rrwick (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nature biotechnology 37, 540–546, https://doi.org/10.1038/s41587-019-0072-8 (2019).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nature Methods 17, 155–158, https://doi.org/10.1038/s41592-019-0669-3 (2020).
Vaser, R. & Šikić, M. Time- and memory-efficient genome assembly with Raven. Nature Computational Science 1, 332–336, https://doi.org/10.1038/s43588-021-00073-4 (2021).
Shafin, K. et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nature biotechnology 38, 1044–1053, https://doi.org/10.1038/s41587-020-0503-6 (2020).
Solares, E. A. et al. Rapid Low-Cost Assembly of the Drosophila melanogaster Reference Genome Using Low-Coverage, Long-Read Sequencing. G3 Genes|Genomes|Genetics 8, 3143–3154, https://doi.org/10.1534/g3.118.200162 (2018).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome research 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Wick, R. R. & Holt, K. E. Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLOS Computational Biology 18, e1009802, https://doi.org/10.1371/journal.pcbi.1009802 (2022).
García-Alcalde, F. et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679, https://doi.org/10.1093/bioinformatics/bts503 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Broad Institute of MIT and Harvard. Picard: A set of command line tools (in Java) for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. https://broadinstitute.github.io/picard (2014).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btac808 (2022).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. bioRxiv, 2023.2011. 2018.567668 (2023).
Xu, W.-D., Lun, Z.-R. & Gajadhar, A. Chromosome numbers of Tritrichomonas foetus and Tritrichomonas suis. Vet Parasitol 78, 247–251, https://doi.org/10.1016/S0304-4017(98)00150-2 (1998).
Zubáčová, Z., Cimbůrek, Z. & Tachezy, J. Comparative analysis of trichomonad genome sizes and karyotypes. Molecular and Biochemical Parasitology 161, 49–54, https://doi.org/10.1016/j.molbiopara.2008.06.004 (2008).
Benchimol, M. et al. Tritrichomonas foetus strain K, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:MLAK00000000.1 (2016).
Senior, E. Tritrichomonas foetusisolate Belfast, whole genome shotgun sequencing project, GenBank, https://identifiers.org/ncbi/insdc:CAJHQR000000000.1 (2021).
Carlton, J. M. et al. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science (New York, N.Y.) 315, 207–212, https://doi.org/10.1126/science.1132894 (2007).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Smith, A., Hubley, R. & Green, P. RepeatMasker Open-4.0. RepeatMasker Open-4.0 (2013).
Du, L., Zhang, C., Liu, Q., Zhang, X. & Yue, B. Krait: an ultrafast tool for genome-wide survey of microsatellites and primer design. Bioinformatics 34, 681–683, https://doi.org/10.1093/bioinformatics/btx665 (2017).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337, https://doi.org/10.1093/bioinformatics/btp157 (2009).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic acids research 49, D192–D200, https://doi.org/10.1093/nar/gkaa1047 (2020).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769, https://doi.org/10.1093/bioinformatics/btv661 (2016).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3, lqaa108, https://doi.org/10.1093/nargab/lqaa108 (2021).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-Genome Annotation with BRAKER. Methods Mol Biol 1962, 65–95, https://doi.org/10.1007/978-1-4939-9173-0_5 (2019).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom Bioinform 2, lqaa026, https://doi.org/10.1093/nargab/lqaa026 (2020).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22, 566, https://doi.org/10.1186/s12859-021-04482-0 (2021).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62, https://doi.org/10.1186/1471-2105-7-62 (2006).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Iwata, H. & Gotoh, O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic acids research 40, e161, https://doi.org/10.1093/nar/gks708 (2012).
Gotoh, O. A space-efficient and accurate method for mapping and aligning cDNA sequences onto genomic sequence. Nucleic acids research 36, 2630–2638, https://doi.org/10.1093/nar/gkn105 (2008).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic acids research 33, 6494–6506, https://doi.org/10.1093/nar/gki937 (2005).
Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic acids research 51, D445–d451, https://doi.org/10.1093/nar/gkac998 (2023).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic acids research 28, 45–48, https://doi.org/10.1093/nar/28.1.45 (2000).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature genetics 25, 25–29, https://doi.org/10.1038/75556 (2000).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic acids research 47, D309–d314, https://doi.org/10.1093/nar/gky1085 (2019).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic acids research 36, 3420–3435, https://doi.org/10.1093/nar/gkn176 (2008).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic acids research 51, D418–D427, https://doi.org/10.1093/nar/gkac993 (2022).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic acids research 48, D498–d503, https://doi.org/10.1093/nar/gkz1031 (2020).
Mölder, F. et al. Sustainable data analysis with Snakemake [version 2; peer review: 2 approved]. F1000Research 10, https://doi.org/10.12688/f1000research.29032.2 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP514276 (2024).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075, https://doi.org/10.1093/bioinformatics/btt086 (2013).
Huang, N. & Li, H. compleasm: a faster and more accurate reimplementation of BUSCO. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btad595 (2023).
Nevers, Y. et al. Quality assessment of gene repertoire annotations with OMArk. Nature biotechnology https://doi.org/10.1038/s41587-024-02147-w (2024).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359, https://doi.org/10.7717/peerj.7359 (2019).
Acknowledgements
The authors thank Prof. Jane Carlton at the Johns Hopkins Bloomberg School of Public Health, Baltimore, USA; and Oliver Rupp and Jochen Blom at the Justus Liebig University Giessen, Germany for their support and valuable discussion. Dr. M. Böhringer and Dr. A. Bernd are thanked for their support with the FACS analysis of T. foetus KV-1. The bioinformatics support of the BMBF-funded project “Bielefeld-Gießen Center for Microbial Bioinformatics” - BiGi (grant 031A533) within the German Network for Bioinformatics Infrastructure (de.NBI) is gratefully acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M.Y.A.-G.: Next Generation Sequencing, bioinformatics, data interpretation and writing of the manuscript, J.S.: Next Generation sequencing, Hi-C sequencing and data analysis; D.W.: Pipeline investigation and data interpretation, H.N.: data interpretation and manuscript revision; L.D.S.: Conceptualisation, project supervision, cultivation of T. foetus KV-1 and DNA preparation; data interpretation, and writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdel-Glil, M.Y., Solle, J., Wibberg, D. et al. Chromosome-level genome assembly of Tritrichomonas foetus, the causative agent of Bovine Trichomonosis. Sci Data 11, 1030 (2024). https://doi.org/10.1038/s41597-024-03818-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03818-8
This article is cited by
-
Whole Genome Sequence of the gut commensal protist Tritrichomonas musculus isolated from laboratory mice
Scientific Data (2025)
-
Chromosome-level genome assembly of Trichomonas vaginalis strain IR-78 (ATCC 50138)
Scientific Data (2025)
