
Abstract
Pygmy jerboas are one of the smallest taxa of rodents. They exhibit distinctly different morphological and biological characteristics from other subfamilies, such as more restricted distribution, species richness, reproductive ability, and population size. Agricultural expansion and the development of new energy projects in recent years lead to sharp decline of their natural populations. Here, we assembled and annotated the first reference genome for the subfamily Cardiocraniinae using Illunima and Nanopore sequencing from the thick-tailed pygmy jerboa, Salpingotus crassicauda. The final genome is 2.44 Gb in size, with a contig N50 length of 13.71 Mb and a BUSCO completeness of 96.35%. A total of 23,344 protein-coding genes were annotated in the final genome. We also determined the mitochondrial genome of this species and annotated 13 protein-coding genes, 22 tRNAs, and 2 rRNA. These genomic assemblies provide resources in studying phylogeny and adaptive evolution of Dipodidae, as well as implementing conservation management of jerboas.
Similar content being viewed by others

Background & Summary
Rodents underwent an extraordinary adaptive radiation throughout the Cenozoic, diverging nearly half of the current mammalian diversity with more than 2,277 species organized into 481 genera1,2. They have occupied almost all terrestrial ecosystems with a wide range of life histories and ecomorphological adaptations including fossorial, arboreal, subaquatic, jumping and gliding capacities2. In deserts, semi-deserts, and steppes of North Africa and Eurasia, a group of bipedal locomotion rodents, jerboas, has successfully evolved and survived in the scarce environments. Jerboas (Dipodidae), belonging to the superfamily of Dipodoidea (Rodentia, Myomorpha), include four closely related phylogenetic groups (Cardiocraniinae, pygmy jerboas; Euchoreutinae, long-eared jerboa; Dipodinae, three-toed jerboas; and Allactaginae, five-toed jerboas)3.
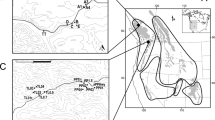
Pygmy jerboas are the smallest group of jerboas and exhibit morphological characteristics distinct from other subfamily species. Their body length is always not exceeding 65 mm, but the head is relatively large that constituting about 40% of the body length (Fig. 1a). The most interesting thing is the heart-shaped skull on account of hypertrophic development of tympanic bullae that strongly broadening the posterior skull section of skull and sharply narrowed anterior section of skull (Fig. 1b)4. The Cardiocraniinae species are distributed in arid landscapes of Mongolia, China, Kazakhstan, Iran, Afghanistan and Baluchistan. However, compared to the two successful subfamilies Allactaginae and Dipodinae, Cardiocraniinae is much more restricted in distribution range, species richness, reproductive potential, and population size. In addition, the position of the phylogenetic relationships of Cardiocraniinae within Dipodidae has been a subject of intense debate due to the lack of available genomic resources.

Morphological feature of S. crassicauda. (a) Hand-drawn view of the external morphology of S. crassicauda; (b) Dorsal view, ventral view of the skull and lateral view of the mandible of S. crassicauda.
In recent years, agricultural expansion and the development of new energy projects (photovoltaic panels and wind farms) in the inland deserts of Asia, especially in China, have caused severe decline in the availability and quality of pygmy jerboas’ habitat, leading to a significant decline of their natural populations (according to our field sampling survey). At the same time, the lack of survey and research data on this group makes it very difficult to assess their conservation status. Genomic resources could provide insights into the phylogeny and evolutionary history, and adaptive evolution of Cardiocraniinae, as well as understand the demographic history and genetic diversity to implement conservation management at natural populations in future5,6. However, thus far, high-quality reference genome of species in Cardiocraniinae has not been assembled.
In this report, we present the first de novo genome assembly and annotation in Salpingotus crassicauda, Cardiocraniinae, Dipodidae, using a hybrid sequencing strategy of long read Nanopore platform (166.25 Gb) and short read Illumina platform (160.28 Gb) (Table 1). This species is mainly distributed in sandy deserts and semideserts of Western and Southern Mongolia, Northwest and Northern China, and extreme east of Kazakhstan. The genome survey revealed an estimated genome size of approximately 2.33 Gb for S. crassicauda. Finally, we generate a 2.44 Gb genome assembly with contig N50 of 13.71 Mb (Table 2). The genome assembly consisted of 1.08 Gb (44.42%) repetitive sequences. A total of 23,344 protein-coding genes were predicted, and 99.05% of them were functionally annotated (Table 3). The availability of the first genomic data and annotation for S. crassicauda represents a valuable genomic resource and provides information for understanding the phylogeny and evolutionary history, adaptive evolution, and conservation needs of Dipodidae.
Methods
Sample collection
In this study, a male S. crassicauda with body weight of 9.8 g was captured on August 13, 2019, in Mori Kazakh Autonomous County, Changji Hui Autonomous Prefecture, Xinjiang Uygur Autonomous Region, China. This individual was euthanasia with a cervical dislocation after ether anesthesia. Six tissues (muscle, heart, liver, spleen, lung, and kidney) were sampled and frozen immediately using liquid nitrogen in the field. An extra muscle tissue preserved in anhydrous ethanol and stored in a −20 °C freezer. All procedures were conducted in accordance with Animal Research Protocol IOZ-2006 approved by the Animal Care Committee of the Institute of Zoology, Chinese Academy of Sciences (IOZCAS).
DNA library construction and genome sequencing
Genomic DNA from muscle tissue was extracted using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). Subsequently, we utilized a combination of Nanopore and Illunima platforms to obtain the genomic sequences of S. crassicauda. (1) For ONT (Oxford Nanopore Technologies) PromethION sequencing, approximately 15 µg of genomic DNA was processed according to the Ligation Sequencing Kit 1D (SQK-LSK109) protocol. After DNA purification and quantifying, the adapters from the LSK109 Ligation kit (cat. SQK-LSK109; Oxford) were used for the ligation reaction, and finally the Qubit 3.0 Fluorometer (Invitrogen) was used to quantify the constructed DNA library. The final 20 kb insert size DNA library was sequenced using Nanopore PromethION platform (Oxford Nanopore, Oxford, UK) in Novogene (Beijing). (2) For Illunima sequencing, genomic libraries with 350-base pair (bp) inserts were constructed with TrueSeq DNA PCR-Free kits (Illumina) following the manufacturer’s instructions. DNA sequencing was performed with 150 bp paired-end reads on Illumina HiSeq platform. The following strategies were used to filter the Illumina raw data: (a) reads with ≥10% unidentified nucleotides (N); (b) reads with adapters; and (c) reads with low-quality bases (≤5) accounting for more than 50% of the total length. Finaly, approximately 166.25 Gb of ONT long read sequences and 160.28 Gb of Illumina short read sequences were generated.
RNA isolation and sequencing
The transcriptome sequences provided necessary gene expression data for the genome sequence annotation. Total RNA was extracted from six tissues, including muscle, heart, liver, spleen, lung, and kidney, using RNeasy Midi Kit (Qiagen, Germany). Each sequencing library was prepared from 1 μg total RNA using TruSeq RNA Sample Preparation v2 Kit (Illumina, San Diego, CA, USA) according to manufacturer’s instruction. Six libraries were paired-end sequenced (2 × 150 bp) on the Illumina HiSeq platform and yielded approximately 50.96 Gb of transcriptome data for all six tissues.
Genome survey and assembly
The genome size and polymorphism information of S. crassicauda was assessed through k-mer analysis using the short reads. Fastp v0.23.4 tool7 was used to perform quality control of reads. Jellyfish v2.2.108 was used to count the frequency of 15–37 bp kmers. These k-mer histograms were uploaded to the GenomeScope 2.09 for an estimation of genome size. The results revealed a predicted haploid genome size of approximate 2.33 Gb, with repetitive rate and heterozygous rate of 17.71% and 0.38%, respectively (Fig. 2a and Figure S1). The de novo genome was first assembled upon ONT long reads using wtdbg2 v2.510. The consensus assembly was then generated after combining the polishing of the Nanopore long reads by Racon11 and 3 rounds polishing of the Illumina short clean reads by Pilon12. The final assembly was 2.44 Gb with contig N50 of 13.71 Mb (Table 2). The statistics of genome assembly were calculated using QUAST v5.2.013 (Figure S2). The GetOrganelle v1.7.7.114 was used to assemble the mitochondrial genome (parameters: -F animal_mt -R 10 -k 21, 45, 65, 85, 105) based on the short-reads. MEGA715 was used for manually adjustment. In the mitochondrial genome, we identified 13 protein-coding genes, 22 tRNAs, and 2 rRNA (Fig. 2b).

Genomics features. (a) A k-mer analysis of S. crassicauda genomics feature; (b) Distribution of annotated genes on mitochondrial genome.
Transposable elements and non-coding RNA annotation
To evaluate the presence of transposable elements (TE), we first constructed a de novo repetitive element database using RepeatModeler v1.0.316. Then, RepeatMasker was performed based on a combination of the de novo database, Repbase, and Dfam database specific to Mammalia. The final TE prediction revealed a 44.42% TE content of the S. crassicauda genome (Table 3), slightly lower than the proportion in other jerboas, such as Dipus sowerbyi (51.17%) and Orientallactaga sibirica (53.02%)17. In TE of S. crassicauda, long terminal repeats (LTR) exhibited the most abundant account (42.71%). Transfer RNA (tRNA) was predicted by tRNAscan-SE v.2.0.618 with default parameters, and ribosome RNA (rRNA) was predicted using RNAmmer v.1.219. Pre-miRNAs and other remaining ncRNAs were searched by Infernal v.1.1.220 based on the Rfam datasets. We identified 10,410 miRNAs, 4,893 tRNAs, 1,232 rRNAs, and 6,338 snRNAs (Table 3).
Gene prediction and functional annotation
Protein-coding genes prediction was conducted through a combination of transcriptome-based, homology-based and ab initio methods. All cleaned RNA-seq data of six tissues were mapped to genome using HISAT2 v2.2.121 and assembled using Trinity v2.10.022. Subsequently, the transcriptome-based prediction was performed using PASA v2.0.223. For homology-based prediction, GeMoMa v1.924 with default settings was used with protein data of four rodent species from GenBank, including Spalax galili (GCF_000622305.1), Rattus norvegicus (GCF_000001895.5), Cricetulus griseus (GCF_000223135.1), and Mus musculus (GCF_000001635.26). For ab initio gene prediction, Augustus v2.725, Geneid v1.426, Genescan27, GlimmerHMM v3.0228 and SNAP29 were used. The longest 1000 transcripts obtained from the transcriptome-based method was used for model training. Finally, the predictions from all three methods were combined using EvidenceModeler v1.1.130. Functional annotation of the gene models included the annotation of functional motifs, domains, and possible related biological processes was conducted via BLASTp searches31 (e-value < 10−5) against the NR (nonredundant protein sequences in NCBI), KEGG (Kyoto Encyclopedia of Genes and Genomes)32 and Gene Ontology (GO)33 databases and retrieved from the results of InterPro34. A total of 23,344 protein-coding genes were predicted, in which 23,123 genes were functionally annotated (Table 3).
Data Records
The Nanopore reads, Illumina reads, and RNA reads for S. crassicauda reported in this paper have been deposited in the Genome Sequence Archive35 (GSA) in National Genomics Data Center36, China National Center for Bioinformation (CNCB)/Beijing Institute of Genomics, Chinese Academy of Sciences with accession numbers CRA01683137 and the NCBI Sequence Read Archive (SRA) with accession numbers SRR29313569-SRR2931357638,39,40,41,42,43,44,45 and Genbank under the accession GCA_040869305.146. The genome assembly and annotation files are available in Figshare47 at https://doi.org/10.6084/m9.figshare.25974952.
Technical Validation
Genome assembly evaluation
Cleaned short reads were mapped to the genome with BWA v0.7.1748 to analyze the mapping rate, coverage and average sequencing depth (Table 2). The percentage of reads mapped in proper pair was 94.83%, and genome coverage rate was 99.66%, indicating a high consistency between the short reads and assembled genome. To further test the accuracy of the single base of the genome, we used Samtools v1.19.149 and Picard v3.1.150 to sort and remove duplicate reads of the BWA mapping result. Then GATK v4.5.0.051 was used to detect the single nucleotide polymorphism (SNP) sites. The heterozygous SNP rate was 0.3121%, and homozygous SNP rate was 0.0009%, indicating highly accuracy of single base. Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.7.152 was used to assess the genome completeness by estimating the percentage of expected single-copy conserved orthologs captured in our draft genomes. The Glires data set (n = 13798) from OrthoDBv10 was used because it is the closest available lineage database to our species. Third-party components Miniprot v0.1353 and HMMER v3.154 within BUSCO v5.7.1 were used to verify assembly completeness and gene prediction. The rest of the settings were default in the BUSCO analysis. To evaluate whether the current assembly has comparable genome completeness, we also compared the BUSCOs of S. crassicauda with other published Dipodidae genomes from GenBank, including Jaculus jaculus (GCF_020740685.1), D. sowerbyi (GCA_036346955.1), O. sibirica (GCA_036365145.1) and O. bullata (GCA_004027895.1). The 96.35% ratio of complete BUSCOs of S. crassicauda was similar to other published genomes of jerboa species (Table 4), which indicated that the genome completeness of our assembly was normal.
Genome synteny
We analyzed the synteny between S. crassicauda and other three published Dipodidae genomes with high quality, including J. jaculus, D. sowerbyi, and O. sibirica. The genome-wide proportions of homology between S. crassicauda and other jerboa species were 55.71% with D. sowerbyi, 61.17% with J. jaculus, and 53.58% with O. sibirica (Table 5). The variation of the homology proportion was within reasonable range and correlated with phylogenetic distance and genome quality. Pairwise aligned transcript sequences between S. crassicauda and two species with chromosomal-level genome were used to perform synteny analysis in MCScanX55. Visualization of comparative syntenic map was carried out using the jcvi.graphics.karyotype module by JCVI v1.4.1456 (Fig. 3).

Synteny of contigs in S. crassicauda from Cardiocraniinae and pseudochromosome in D. sowerbyi and J. jaculus from Dipodinae. The segment length shown here does not represent the true chromosome length.
Code availability
No custom scripts or code were used in this study.
References
Wilson, D. E., Mittermeier, R. A. & Lacher, T. E. Handbook of the Mammals of the World (Lynx Edicions, 2019).
Fabre, P. H., Hautier, L., Dimitrov, D. & Douzery, E. J. P. A glimpse on the pattern of rodent diversification: a phylogenetic approach. BMC Evol. Biol. 12, 88, https://doi.org/10.1186/1471-2148-12-88 (2012).
Michaux, J. & Shenbrot, G. in Handbook of the mammals of the world-volume 7: Rodents II (eds. Wilson, D. E., Lacher, T. E. & Mittermeier, R. A.) (Lynx Edicions, 2019).
Shenbrot, G., Sokolov, V., Heptner, V. & Koval’skaya, Y. Jerboas: Mammals of Russia and adjacent regions (CRC Press, 2008).
Xie, H. et al. Ancient demographics determine the effectiveness of genetic prging in endangered lizards. Mol. Biol. Evol. 39, 1537–1719, https://doi.org/10.1093/molbev/msab359 (2021).
Wang, P. et al. Genomic consequences of long-term population decline in brown eared pheasant. Mol. Biol. Evol. 38, 263–273, https://doi.org/10.1093/molbev/msaa213 (2020).
Chen, S. Ultrafast one-pass fastq data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107, https://doi.org/10.1002/imt2.107 (2023).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods. 17, 155–158, https://doi.org/10.1038/s41592-019-0669-3 (2020).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Walker, B. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075, https://doi.org/10.1093/bioinformatics/btt086 (2013).
Jin, J. J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241, https://doi.org/10.1186/s13059-020-02154-5 (2020).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0. 2008–2015, www.repeatmasker.org (2008–2015).
Cheng, J. L. et al. Similar adaptative mechanism but divergent demographic history of four sympatric desert rodents in Eurasian inland. Communciations Biol. 6, 33, https://doi.org/10.1038/s42003-023-04415-y (2023).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57, https://doi.org/10.1093/nar/gkw413 (2016).
Huang, Y., Gilna, P. & Li, W. Z. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25, 1338–1340, https://doi.org/10.1093/bioinformatics/btp161 (2009).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337, https://doi.org/10.1093/bioinformatics/btp157 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19, 189, https://doi.org/10.1186/s12859-018-2203-5 (2018).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Parra, G., Blanco, E. & Guigó, R. GeneID in Drosophila. Genome Res. 10, 511–515, https://doi.org/10.1101/gr.10.4.511 (2000).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94, https://doi.org/10.1006/jmbi.1997.0951 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids res. 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Mitchell, A. L. et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360, https://doi.org/10.1093/nar/gky1100 (2018).
Chen, T. T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genom. Proteom. Bioinf. 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Xue, Y. B. et al. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38, https://doi.org/10.1093/nar/gkab951 (2022).
CNCB Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA016831 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313569 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313570 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313571 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313572 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313573 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313574 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313575 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29313576 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_040869305.1 (2024).
Cheng, J. Genome assembly and annotation of Salpingotus crassicauda. figshare https://doi.org/10.6084/m9.figshare.25974952 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Broad Institute, GitHub Repository. Picard Toolkit. https://broadinstitute.github.io/picard/ (2019).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014, https://doi.org/10.1093/bioinformatics/btad014 (2023).
Eddy, S. R. Accelerated profile HMM searches. PLOS Comput. Biol. 7, e1002195, https://doi.org/10.1371/journal.pcbi.1002195 (2011).
Wang, Y. P. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Tang, H. et al. JCVI: A versatile toolkit for comparative genomics analysis. iMeta. 3, e211, https://doi.org/10.1002/imt2.211 (2024).
Acknowledgements
This research was supported by grants from the Third Xinjiang Scientific Expedition Program (Grant No.2022xjkk0205 to Lin Xia, No.2021xjkk0604 to Jilong Cheng), the National Natural Science Foundation of China (32370472 to Jilong Cheng, 32170416 to Qisen Yang), the Joint Fund of National Natural Science Foundation of China (U2003203 to Lin Xia), and the Key Laboratory of Zoological Systematics and Evolution of the Chinese Academy of Sciences (2023IOZ0104 and Y229YX5105 to Deyan Ge).
Author information
Authors and Affiliations
Contributions
J.C. and L.X. designed and conceived the project. J.C., X.P. and L.X. collected samples and collected field data. J.C., Y.Z. and F.Y. performed the genomic analyses and interpreted the results. J.C., Y.Z., X.P. drafted and prepared original manuscript. F.Y., D.G. and Q.Y. reviewed and edited. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, J., Zhang, Y., Xia, L. et al. De novo genome of thick-tailed pygmy jerboa Salpingotus crassicauda, Cardiocraniinae, Dipodidae. Sci Data 11, 1041 (2024). https://doi.org/10.1038/s41597-024-03905-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03905-w
