
Abstract
Soil microbes are key drivers of ecosystem processes promoting nutrient cycling, system productivity, and resilience. While much is known about the roles of microbes in established systems, their impact on soil development and the successional transformation over time remains poorly understood. Here, we provide 67 diverse, rhizosphere-associated Pseudomonas draft genomes from an undisturbed salt march primary succession spanning >100 years of soil development. Pseudomonas are cosmopolitan bacteria with a significant role in plant establishment and growth. We obtained isolates associated with Limonium vulgare and Artemisia maritima, two typical salt marsh perennial plants with roles in soil stabilization, salinity regulation, and biodiversity support. We anticipate that our data, in combination with the provided physiochemical measurements, will help identify genomic signatures associated with the different selective regimes along the successional stages, such as varying soil complexity, texture, and nutrient availability. Such findings would advance our understanding of Pseudomonas’ role in natural soil ecosystems and provide the basis for a better understanding of the roles of microbes throughout ecosystem transformations.
Similar content being viewed by others

Background & Summary
Advancements in cell culture, DNA extraction, and sequencing technologies have greatly increased our understanding of the variable complexity and diversity of soil microbial communities. Latest technologies enable the collection of samples in situ and provide information on the gene level of the soil microorganisms, which can then be used to infer their ecological roles. One aspect that has interested microbial ecologists is understanding the genomic modifications that allow soil microorganisms to colonize soils during soil development. However, the ability to sample over time spans necessary for soil formation is challenging1. Primary succession or chronosequence have been identified as valuable playgrounds to address these questions due to their associated space-for-time substitution. As such, they have been used to study soil development and functional changes related to the variation in above-ground and below-ground processes2,3,4 providing critical information over temporal community dynamics and soil development across multiple timescales5,6. From a microbial perspective, several researchers have utilized chronosequence to study soil communities in different areas, such as grassland3, forests4, deglaciated soils5, and salt marshes7.
Salt marshes are tidal wetlands that play a vital ecological role in the coastal ecosystem and maintain water quality and habitat health by filtering pollutants, runoff, and excess nutrients. Moreover, they store carbon at a rate ten times that of mature tropical forests, helping moderate climate change effects8. The salt marsh located on the Waddensea barrier island of Schiermonnikoog constitutes a well-documented chronosequence covering more than one hundred years of succession9. Recent studies revealed that soil microbial communities change in taxonomic composition throughout this chronosequence, with soil organic matter and salt concentrations being the major drivers of soil microbial community structure10. These soil microbial communities also differ functionally and are enriched in genes associated with dispersal at the early stages. In contrast, the late stages exhibit enrichment in antibiotic resistance genes5. While these studies have provided important insights into the effects of succession and associated changes in soil biotic and abiotic parameters at the community level, inter- and intraspecies variation remains poorly understood.
Pseudomonas is one of the most studied and diverse bacterial genera because of its prevalence in several environments, such as soil11 and water12, and hosts, such as plants13, mammals14, and insects15. Pseudomonas is ecologically well-studied in soil under anthropogenic influence and is regarded as a key indicator species to reflect disturbances. Pseudomonas genomes have been shown to be highly diverse and adaptable due to their potential for genomic variation16. The diversity and structure of Pseudomonas communities significantly correlate with the changes in soil fertilization17 and the long-term use of mineral fertilizers18. Moreover, the genus Pseudomonas contains several plant-associated and free-living beneficial strains commonly found in soils from sustainable crop production, where they can act as plant growth-promoting or biocontrol agents19. However, research thus far has focused on cultivated soils, while Pseudomonas isolates from lesser explored niches show beneficial properties and genetic heterogeneity20, clearly indicating an untapped potential.
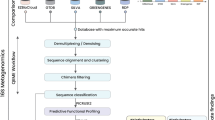
Here, we used the genus Pseudomonas as a focus group to determine how the changes in soil characteristics along the primary succession of Schiermonnikoog influence the selection pressure imposed in this group. Specifically, we aimed to collect data to facilitate the analysis of inter- and intraspecies diversity of different Pseudomonas genotypes colonizing the different niches of the salt marsh soils. By analyzing the genomic diversity within the Pseudomonas genus across different stages of soil development, our study leverages the natural environmental gradient provided by the chronosequence to uncover how microbial communities are shaped by both temporal and environmental factors. For that, we isolated Pseudomonas strains from the endo- and rhizosphere of typical salt marsh plants with broad distribution along the chronosequence21. We provide whole genome sequencing data of 67 Pseudomonas and the physiochemical parameters from the soil samples collected from the roots of salt marsh plants on the island of Schiermonnikoog, the Netherlands. These data represented snapshots of the Pseudomonas community spanning a timescale of 100 years, with successional ages ranging from 5 to 105 years (Fig. 1). From each location, we sampled rhizosphere soil and root endosphere from two plants, Limonium vulgare and Artemisia maritima. The complete data contains the raw sequencing reads, the cleaned assemblies with the taxonomic information collected through sampling, measurement, cultivation of individual isolates, DNA extraction, whole genome sequencing, and the subsequent refinement and annotation steps (Fig. 2). We expect that these data will help study microbial questions from the perspective of gene level, including, but not limited to, microbiology, microbial ecology, genetics, and evolution. In addition, the associated physiochemical parameters, in particular, might link the microbial world and environmental disturbances.

Sampling locations along the chronosequence on the island of Schiermonnikoog. Map of Schiermonnikoog displaying the location where whole genomes were collected from the indicated soil types. Wadden islands like Schiermonnikoog constantly migrate, with soil washed away from the West end and new sand continuously deposited at the East end. Our sampling locations (red bold arrows) are part of a well-documented chronosequence representing more than 100 years of soil development (long dark arrow). The soil stages by years of soil formation are indicated above the location symbols. The type of vegetation habitats is indicated by dark arrows and text below the symbols of locations. This Figure was completed on ArcGis Online, and the base map was obtained from Esri Nederland41.

Overview of sampling procedure, sample processing, genome sequencing, and bioinformatic analysis. Diagram illustrating the main stages and procedures for generating the dataset of this study: endo- and rhizosphere sampling, strain isolation, whole-genome sequencing, and assembly and bioinformatic analysis. Summary metrics of all genomes are provided in the bottom-left box. The figure was created with BioRender.com.
Methods
Plants and root-attached soil were collected in April 2016 from five sites along a successional chronosequence on the Dutch Wadden island Schiermonnikoog (soil ages of 5, 15, 35, 65, and 105 years)21. Within each plot, four healthy-looking L. vulgare and A. maritima of similar sizes with soil adhering to the intact roots were obtained and processed together, generating two composite samples per plot. Thirty composite samples in total were collected (5 stages × 3 plots per stage × 2 plant species). Each sample was placed in a sterile plastic bag, sealed, and transported to the laboratory within 24 hours.
Bulk soil samples were subjected to the measurements of pH, soil water content (SWC), soil organic matter (SOM), nitrate (N-NO3−), ammonium(N-NH4+), soil exchangeable elements (Na, Mg, Ca, and K), phosphate (P), and total nitrogen (TN). Soil chemical analyses were carried out in collaboration with the Department of Community and Conservation Ecology, University of Groningen (see Wang et al. 2016 for a detailed description of the methods)21. Soil physical data (sand: silt: clay % content) were obtained from a previous study carried out in the same sampled sites (see Dini-Andreote et al.)22.
From each composite sample, we sampled rhizosphere soil and root endosphere. For each rhizosphere sample, 8–10 g of soil was diluted in 47 ml 1 × PBS solution, shaken for 30 min at 200 rpm at room temperature, and prepared for serial dilutions (1/10) in sterile 1X PBS. For endosphere samples, the roots were washed under running water, trimmed to remove adhering soil and dead tissues, and surface-sterilized23. The samples were then diced with a sterile scalpel and immersed into 45 ml 0.9% NaCl solution. After incubation for 1 h at 28 °C, the suspension was shaken using a horizontal vortex instrument, followed by serial dilution (1/10) in sterile 1X PBS. Sterility checks were performed by tissue-blotting surface-sterilized root samples on R2A plates at 28 °C for 2–7 days. Only samples without bacterial growth were considered successfully sterilized and used further.
Pseudomonas isolates were obtained using Gould’s S1 medium, a selective medium previously used for isolating fluorescent Pseudomonas24. Thirty-two bacterial colonies per plate with unique morphologies were purified using a streak-plate procedure, transferred onto new S1 and R2A25 medium plates, and further used as templates for BOX-PCR, which is a DNA-based typing method capable of simultaneously screening many DNA regions scattered in the bacterial genome26. 109 bacterial cultures with unique BOX-PCR patterns on Gould’s S1 agar plates were subjected to total DNA extraction using the MoBio UltraClean Microbial DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA). All DNA samples were standardized to an equal concentration of 5 ng μL−1 for sequencing. 70 Pseudomonas spp unique isolates were sent to LGC Genomics GmbH (Berlin, Germany) for whole genome sequencing on an Illumina NextSeq. 500 V2 platform with a 150-bp paired-end library design.
We trimmed the resulting Illumina reads by using Trim Galore27, then assembled the trimmed reads de novo with SPAdes28, aiming for genome sizes around 6 Mbp with GC contents of 60%–70%29. (Pseudomonas aeruginosa 65–67%, size 5.5–7 Mbp; Pseudomonas fluorescens ~60%, size ~6 Mbp30). We kept only scaffolds larger than 1000 bp (Table S1) and further decontaminated the raw contigs to obtain high-quality draft genomes (see Technical Validation for more information). We performed taxonomic classification using the Genome Taxonomy Database (GTDB) and the associated toolkit (GTDBtk)31. Individual species were demarcated at a threshold of less than 95% of average nucleotide identity (ANI)32. Detailed taxonomic information is provided in Table S4 and assigned species names in Table S5. To validate the positioning of the strains not assigned to species level by GTDBtk into the genus Pseudomonas, we reconstructed a phylogenetic tree (Fig. 4). We used 34 reference genomes, including 9 outgroup species and the 25 closest reference genomes identified by GTDBtk (NCBI genome accession listed in Table S5). We used GTDBtk to extract 120 universal bacterial marker amino acid alignments from all genomes and FastTree33 to reconstruct a phylogenetic tree based on the amino acid sequence alignments. The resulting tree was visualized with ggtree34 and Evolview35. The overview of the procedures is displayed in Fig. 2.
Data Records
Physiochemical parameters can be found in Table 1. The raw Illumina sequencing reads have been deposited in the European Nucleotide Archive (ENA) under the accession number ERP14230636. The curated and annotated assemblies for this study have been deposited in NCBI GenBank under the accessions GCA_96397[0405–1065]37. The detailed accession numbers for each samples, library size, and coverage statistics can be found in Table S5.
Technical Validation
The overall sequencing quality of the short-read data was assessed with FastQC38 and low-quality regions were trimmed with Trim Galore27. To ensure high-quality genomes, assemblies were manually screened for low-coverage contamination and suspicious contigs were removed. In total, we recovered 67 genomes with good quality based on CheckM39 metrics (Completeness > 80%, contamination > 10%, completeness – 5*contamination > 80%) (Fig. 3).

In-silico decontamination considerably improves Pseudomonas genome assembly quality. Quality metrics of genome assemblies before (orange) and after (green) decontamination: the size of the longest contig, number of scaffolds, completeness, contamination, and integrated score calculated as completeness - 5*contamination. All values were computed with CheckM39. Quality improvements indicate that manual decontamination based on sequence coverage and length removed contaminations efficiently without affecting assembly completeness.

Phylogenetic relationships of 67 Pseudomonas isolates and their distribution across soil stages, host plants, and habitat. Phylogenetic tree including nine outgroup species (first 9 species from the bottom), 26 Pseudomonas reference genomes from the same GTDB subgroup E as our samples, and the 67 Pseudomonas genomes of this study. New genomes were assigned to known species based on closest placement in the Genome Taxonomy Database (GTDB)29. 31 genomes did not match at species-level cutoff (ANI > = 95%, AF > = 0.5) to known Pseudomonas and can be considered candidates for new species. Isolates were obtained from 5 different soil stages (5 years to 105 years), from two host plants (Artemisia maritima and Limonium vulgare) and from both endo- and rhizosphere.
Code availability
Software versions and any relevant variables and parameters employed are as follows:
• Trim Galore v0.6.5 (trim_galore –paired –fastqc --phred33 --illumina)27;
• FastQC v0.11.938;
• SPAdes v3.15.4 (spades.py --careful -t 20)28;
• Seqkit v2.3.0 (seqkit grep -f)40;
• CheckM v1.1.3-foss-2021a (checkm lineage_wf -x fa)39;
• GTDB v 1.7.0 (gtdbtk classify_wf); GTDB-Tk reference data version r20231;
• FastTree v2.1.1133;
• ggtree v3.9.134.
References
Chapin, F. S., Walker, L. R., Fastie, C. L. & Sharman, L. C. Mechanisms of Primary Succession Following Deglaciation at Glacier Bay, Alaska. Ecol. Monogr. 64, 149–175 (1994).
García Hernández, E., Berg, M. P., Van Oosten, A. R., Smit, C. & Falcão Salles, J. Linking Bacterial Communities Associated with the Environment and the Ecosystem Engineer Orchestia gammarellus at Contrasting Salt Marsh Elevations. Microb. Ecol. 82, 537–548 (2021).
Schrama, M. J. J., Plas, F., van der, Berg, M. P. & Olff, H. Decoupled diversity dynamics in green and brown webs during primary succession in a saltmarsh. J. Anim. Ecol. 86, 158–169 (2016).
Salles, J. F. et al. Successional patterns of key genes and processes involved in the microbial nitrogen cycle in a salt marsh chronosequence. Biogeochemistry 132, 185–201 (2017).
Dini-Andreote, F., van Elsas, J. D., Olff, H. & Salles, J. F. Dispersal-competition tradeoff in microbiomes in the quest for land colonization. Sci. Rep. 8, 9451 (2018).
Wang, M., Li, E., Liu, C., Jousset, A. & Salles, J. F. Functionality of root-associated bacteria along a salt marsh primary succession. Front. Microbiol. 8, (2017).
Dini-Andreote, F., Pylro, V. S., Baldrian, P., van Elsas, J. D. & Salles, J. F. Ecological succession reveals potential signatures of marine–terrestrial transition in salt marsh fungal communities. ISME J. 10, 1984–1997 (2016).
Santos, P. X. Alicia Wilson, Zhenming Ge, Isaac. Understanding the Importance of Salt Marshes. Eos http://eos.org/editors-vox/understanding-the-importance-of-salt-marshes (2022).
Olff, H., De Leeuw, J., Bakker, J. P., Platerink, R. J. & van Wijnen, H. J. Vegetation Succession and Herbivory in a Salt Marsh: Changes Induced by Sea Level Rise and Silt Deposition Along an Elevational Gradient. J. Ecol. 85, 799–814 (1997).
Dini-Andreote, F., Brossi, M. J. de L., van Elsas, J. D. & Salles, J. F. Reconstructing the Genetic Potential of the Microbially-Mediated Nitrogen Cycle in a Salt Marsh Ecosystem. Front. Microbiol. 7, (2016).
Butaitė, E., Baumgartner, M., Wyder, S. & Kümmerli, R. Siderophore cheating and cheating resistance shape competition for iron in soil and freshwater Pseudomonas communities. Nat. Commun. 8, 414 (2017).
Mulet, M. et al. Pseudomonas Species Diversity Along the Danube River Assessed by rpoD Gene Sequence and MALDI-TOF MS Analyses of Cultivated Strains. Front. Microbiol. 11, 2114 (2020).
Shalev, O., Ashkenazy, H., Neumann, M. & Weigel, D. Commensal Pseudomonas protect Arabidopsis thaliana from a coexisting pathogen via multiple lineage-dependent mechanisms. ISME J. 16, 1235–1244 (2022).
Zhou, W. et al. Symbiotic bacteria mediate volatile chemical signal synthesis in a large solitary mammal species. ISME J. 15, 2070–2080 (2021).
Saati-Santamaría, Z. et al. Discovery of Phloeophagus Beetles as a Source of Pseudomonas Strains That Produce Potentially New Bioactive Substances and Description of Pseudomonas bohemica sp. nov. Front. Microbiol. 9, 913 (2018).
Silby, M. W., Winstanley, C., Godfrey, S. A. C., Levy, S. B. & Jackson, R. W. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680 (2011).
Su, X. et al. Long-term organic fertilization changes soil active bacterial composition and multifunctionality: RNA-based bacterial community and qPCR-based SmartChip analysis. J. Soils Sediments 21, 799–809 (2021).
Tambong, J. T. & Xu, R. Culture-independent analysis of Pseudomonas community structures in fertilized and unfertilized agricultural soils. Ann. Microbiol. 63, 323–333 (2013).
El-Saadony, M. T. et al. Plant growth-promoting microorganisms as biocontrol agents of plant diseases: Mechanisms, challenges and future perspectives. Front. Plant Sci. 13, (2022).
Sah, S. & Singh, R. Phylogenetical coherence of Pseudomonas in unexplored soils of Himalayan region. 3 Biotech 6, 170 (2016).
Wang, M., Yang, P. & Falcão Salles, J. Distribution of Root-Associated Bacterial Communities Along a Salt-Marsh Primary Succession. Front. Plant Sci. 6, (2016).
Dini-Andreote, F. et al. Dynamics of bacterial community succession in a salt marsh chronosequence: evidences for temporal niche partitioning. ISME J. 8, 1989–2001 (2014).
Sahu, P. K. et al. Surface sterilization for isolation of endophytes: Ensuring what (not) to grow. J. Basic Microbiol. 62, 647–668 (2022).
Gould, W. D., Hagedorn, C., Bardinelli, T. R. & Zablotowicz, R. M. New Selective Media for Enumeration and Recovery of Fluorescent Pseudomonads from Various Habitats. Appl. Environ. Microbiol. 49, 28 (1985).
Ellis, R. J., Morgan, P., Weightman, A. J. & Fry, J. C. Cultivation-Dependent and -Independent Approaches for Determining Bacterial Diversity in Heavy-Metal-Contaminated Soil. Appl. Environ. Microbiol. 69, 3223–3230 (2003).
Brusetti, L. et al. Fluorescent-BOX-PCR for resolving bacterial genetic diversity, endemism and biogeography. BMC Microbiol. 8, 220 (2008).
Krueger, F. et al. FelixKrueger/TrimGalore. doi:0.5281/zenodo.5127898 (2023).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinforma. 70, (2020).
Hesse, C. et al. Genome-based evolutionary history of Pseudomonas spp. Environ. Microbiol. 20, 2142–2159 (2018).
Klockgether, J., Cramer, N., Wiehlmann, L., Davenport, C. F. & Tümmler, B. Pseudomonas aeruginosa Genomic Structure and Diversity. Front. Microbiol. 2, 150 (2011).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics btac672, https://doi.org/10.1093/bioinformatics/btac672 (2022).
Grandcolas, P. et al. Mapping characters on a tree with or without the outgroups. Cladistics 20, 579–582 (2004).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLOS ONE 5, e9490 (2010).
Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinforma. 69, e96 (2020).
Subramanian, B., Gao, S., Lercher, M. J., Hu, S. & Chen, W.-H. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 47, W270–W275 (2019).
Siyu, M., Miao, W., Joana, F. S. & Thomas, H. Whole genome shotgun sequencing of Pseudomonas from a soil chronosequence. ENA Read archive https://identifiers.org/ena.embl:ERP142306 (2023).
Mei, S., Wang, M., Salles, J. F. & Hackl, T. Draft genomes of Pseudomonas from a soil chronosequence. GeneBank https://identifiers.org/ncbi/insdc.gca:GCA_96397[0405-1065] (2023).
Andrew, S. FastQC: A Quality Control tool for High Throughput Sequence Data [Online]. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLOS ONE 11, e0163962 (2016).
Jan Willem va Aalst. Esri Nederland. www.imergis.nl.
Acknowledgements
We thank Han Olff, Matty Berg, Chris Smit, Maarten Schrama, and Ruth Howison for information on sampling locations and plant species. We are grateful to Jolanda K. Brons and Armando Cavalcante Franco Dias for sampling expeditions. We thank the’Nederlandse Vereniging voor Natuurmonumenten’ for granting us access to the salt marsh. We thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine and Hábrók high-performance computing cluster. We thank the China Scholarship Council (CSC), on a personal grant to SM (Grant No. [2020]596) and MW (Grant No. [2013]3009).
Author information
Authors and Affiliations
Contributions
M.W. and J.F.S. designed the study. M.W. carried out the field and lab work. S.M., J.F.S. and T.H. designed the data analysis. S.M. carried out the data processing and analysis with contributions from T.H. S.M. and T.H. wrote the manuscript with contributions from J.F.S. J.F.S. and T.H. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mei, S., Wang, M., Salles, J.F. et al. Diverse rhizosphere-associated Pseudomonas genomes from along a Wadden Island salt marsh transition zone. Sci Data 11, 1140 (2024). https://doi.org/10.1038/s41597-024-03961-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03961-2
