
Abstract
The family Pyralidae is a large and important group within the superfamily Pyraloidea, consisting of more than 5,900 described species. The rice moth Corcyra cephalonica is a noteworthy pest affecting stored grains and products. However, the absence of high-quality genomes hinders our comprehension of the ecology of C. cephalonica. Herein, we propose a chromosome-level genome of C. cephalonica achieved through integrating Nanopore, Illumina, and Hi-C reads. The genome of 453.37 Mb in size consists of 98 scaffolds and 154 contigs, with a scaffold N50 size of 16.67 Mb. The majority of contigs (99.36%, 450.45 Mb) were effectively anchored to 29 chromosomes. The BUSCO analysis of the assembly revealed a completeness of 99.0% (n = 1,367), identifying 98.6% single-copy BUSCO genes and 0.4% duplicated BUSCO genes. Furthermore, the genome contains 48.74% (220.96 Mb) repeat elements and encompasses 14,135 predicted protein-coding genes. This study offers a valuable genomic foundation for advancing our comprehension of the biology of C. cephalonica.
Similar content being viewed by others
Background & Summary
The family Pyralidae, belonging to the superfamily Pyraloidea, is a significant and expansive group within the Lepidoptera order, encompassing more than 5,900 described species1. This family exhibits greater diversity in Asia and Africa than in the Western Hemisphere2. While most Pyralidae larvae are phytophagous, the larvae of some species are notorious pests of various crops, such as sugarcane, corn, and rice, as well as stored products, including food, flour, seeds, grains, and fodder3. The rice moth Corcyra cephalonica (Stainton, 1886) is a Pyralidae pest known for infesting stored grains and products. This insect pest has been documented on various continents, including Europe, Africa, Asia, and North America4, and its spread to other regions is facilitated by the transport of infested grains and commodities5. The larval stage of C. cephalonica is particularly destructive and tends to infest damaged grains. As feeding progresses, the larvae produce dense silken fibers that form lumps within the infested products5. The presence of larval feces, webbings, exuviae, and cadavers contaminates the products, reducing consumer acceptance and market value6. In addition to its detrimental role as a pest, C. cephalonica acts as a laboratory host for various biocontrol agents, including Trichogrammatoidea eldanae (Hymenoptera: Trichogrammatidae), and Habrobracon hebetor (Hymenoptera: Braconidae)7,8,9. Nevertheless, the absence of a high-quality genome substantially impedes our comprehension of the ecological facets of C. cephalonica.
This study proposes assembling a genome of C. cephalonica by integrating data from Oxford Nanopore Technologies (ONT), Illumina, and Hi-C. Our analysis included annotations for repeats, non-coding RNAs, and protein-coding genes. The genome of C. cephalonica offers a valuable genomic foundation for conducting ecological and functional studies.
Methods
Sample collection and sequencing
A male specimen of C. cephalonica was obtained for sequencing on June 28, 2022, in Nanjing, China. To mitigate the risk of external contaminants, the sample underwent a rigorous washing process with a phosphate-buffered saline solution for ten minutes. Subsequently, the samples were promptly frozen using liquid nitrogen for 20 minutes and subsequently kept in the laboratory at −80 °C until the sequencing process.
The genomic DNA was isolated from the specimens using the FastPure® Blood/Cell/Tissue/Bacteria DNA Isolation Mini Kit produced by Vazyme Biotech Co., Ltd., located in Nanjing, China. TRIzol reagent from YiFeiXue Tech in Nanjing, China, was used for RNA extraction. The libraries were prepared for Illumina sequencing using the TruSeq DNA PCR-free Kit, resulting in paired-end reads of 150 bp. The Hi-C experiment was carried out following a previously published protocol10. The short-read library sequencing data were generated on the Illumina NovaSeq6000 platform. A 30 kb library was created using the SQK-LSK109 1D DNA Ligation Sequencing Kit on the ONT PromethION platform. Berry Genomics (Beijing, China) carried out the library construction and sequencing procedures. Our sequencing efforts yielded 148.52 Gb of high-quality data. This dataset comprised 41.92 Gb (92.47×) of long reads from ONT sequencing, 39.52 Gb (87.16×) of short reads from Illumina sequencing, 56.27 Gb (124.12×) of Hi-C data, and 10.81 Gb of transcriptome reads (Table 1).
Genome assembly
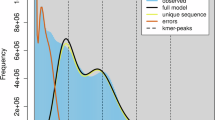
The Illumina data underwent quality control using BBTools v38.8211. The “clumpify.sh” script was utilized to remove duplicate reads. Furthermore, the “bbduk.sh” script was used to trim the reads to a stringent standard. The process involved filtering out sequences with quality scores below 20 and removing sequences containing more than 5 Ns. To estimate the size, heterozygosity, and content of repetitive sequences in the C. cephalonica genome, we performed a genome survey utilizing GenomeScope v2.012. The estimated genome size was between 422.17 and 423.22 Mb, displaying a high heterozygosity rate (1.04%–1.07%) (Fig. 1).

GenomeScope genome size estimates for Corcyra cephalonica.
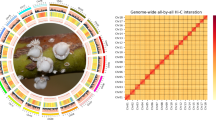
The initial assembly of ONT reads was performed using Nextdenovo v2.3.113 with the default parameters. Subsequently, the primary assembly underwent two rounds of polishing using Illumina reads with NextPolish v1.3.114. Purge_Dups v1.2.515 was utilized to eliminate heterozygous regions with a haploid cutoff value of 60 (‘-s 60’). The Hi-C reads underwent quality control procedures before being aligned to the assembly using Juicer v1.6.216. Subsequently, we employed 3D-DNA v.18092217 to anchor contigs and construct chromosomes. For assembly accuracy validation, a comprehensive examination of the contig assembly outcomes was performed, with manual correction of any assembly errors executed through Juicebox v.1.11.0816. In our efforts to identify potential contaminants within the genome assembly, we employed MMseqs2 v1118 for searches against the NCBI nucleotide and UniVec databases. Furthermore, we utilized BLAST + v2.11.019 to identify vector contaminants by cross-referencing with the UniVec database. The sequences matching over 90% with the specified databases were identified as potential contaminants. Moreover, sequences displaying over 80% similarity underwent thorough validation via BLASTN analysis against the NCBI nucleotide database. The bacterial and fungal contaminants were systematically removed from the scaffolds based on the search results. In the process of identifying autosomes and sex chromosomes, we employed MiniMap2 v2.1720 to align both the raw ONT reads and Illumina reads to the assembly. Following this, the coverage of each chromosome was meticulously calculated using SAMtools v1.921. Notably, one chromosome exhibited approximately half the coverage in comparison to the others, which is indicative of the Z chromosome. The final assembly of the C. cephalonica genome measures 453.37 Mb in size, composed of 98 scaffolds and 154 contigs. Notably, the scaffold N50 size reached 16.67 Mb, while the contig N50 size amounted to 12.61 Mb (Table 2). Most contigs (99.36%, 450.45 Mb) were successfully anchored to 29 chromosomes, ranging in length from 10.43 to 52.19 Mb (Table 3; Figs. 2, 3). The chromosome coverage data has been calculated and is outlined in Table 3. Particularly noteworthy is Chromosome 1, which displayed an ONT long-read sequence coverage of 52.68 and an Illumina short-read sequence coverage of 38.61. This coverage profile is notably distinctive, being approximately half that of the other chromosomes in the analysis. As a result, chromosome 1 was definitively designated as the Z chromosome (Table 3).

Genome-wide chromosomal heatmap of Corcyra cephalonica, where each chromosome is delineated in blue, and each contig is highlighted with a green outline.

The Genome characteristics of Corcyra cephalonica are depicted in a circular layout. Starting from the outer ring to the inner ring, the distributions include chromosome length, GC content, gene density, and various genomic features such as transposable elements (DNA, short interspersed nuclear elements (SINE), long interspersed nuclear elements (LINE), and long terminal repeats (LTR)), as well as simple repeats.
Genome annotation
A de novo repeat library specific to C. cephalonica was generated using RepeatModeler v2.0.422. This particular repeat library was subsequently integrated with RepBase-2013090923 and Dfam 3.524, culminating in the creating of a customized repeat library. We utilized RepeatMasker v.4.1.425 to identify and mask repetitive elements present in the C. cephalonica genome by aligning it with a custom repeat library. The analysis performed by RepeatMasker revealed that approximately 48.74% of the C. cephalonicas genome consists of repetitive sequences, encompassing unclassified elements (11.70%), LINE transposons (12.11%), SINE transposons (7.24%), DNA transposons (3.04%), LTR transposons (2.29%), and other types of elements (Table 4).
The identification of non-coding RNAs (ncRNAs) in C. cephalonica was executed using Infernal v1.1.426 based on the Rfam v14.10 database27. Additionally, transfer RNAs were detected through the utilization of tRNAscan-SE v2.0.928. The analysis unveiled a diverse repertoire of 679 ncRNAs in the C. cephalonicas genome, comprising 83 small nuclear RNAs, 77 microRNAs, and 334 tRNAs (Table 4).
Protein-coding gene annotation in the C. cephalonica genome was performed using MAKER v3.01.0329. The annotation process seamlessly integrated data from the transcriptome, ab initio predictions, and homologous protein evidence. To align the transcribed RNA, HISAT2 v2.2.130 was used, and the resulting RNA-seq alignment was used for genome-guided assembly with StringTie v2.1.631. In the ab initio predictions, BRAKER v2.1.632 was employed, utilizing GeneMark-ES/ET/EP 4.68_lic33 and Augustus v3.4.034, which were automatically trained to incorporate transcriptome sequences and proteins sourced from the OrthoDB v11 database35. Furthermore, GeMoMa v1.936 was employed for gene prediction based on protein homology, utilizing protein sequences sourced from five distinct species: Drosophila melanogaster (GCF_000001215.4)37, Bombyx mori (GCF_014905235.1)38, Pieris rapae (GCF_905147795.1)39, Spodoptera frugiperda (GCF_011064685.2)40, and Chilo suppressalis (GCA_902850365.2)41. The analysis yielded a prediction of 14,135 protein-coding genes within the C. cephalonica genome, each averaging a length of 10,539.5 bp (Table 4). BUSCO was used to assess the completeness of the protein sequences, which achieved a high score of 99.3% (n = 1,367). This included 89.2% (1,219) single-copy, 10.1% (138) duplicated, 0.6% (8) missing, and 0.1% (2) fragmented BUSCOs, affirming the accuracy and reliability of the gene predictions.
We conducted thorough analyses against the UniProtKB database utilizing Diamond v2.0.1142 for gene functional annotation searches. In addition, we utilized eggNOGmapper v2.0.143 and InterProScan 5.53–87.044 to annotate Gene Ontology, identify KEGG pathways, and explore Reactome pathways. The InterProScan analyses encompassed five databases: Pfam45, SMART46, Superfamily47, Gene3D48, and CDD49. Functional annotation revealed that C. cephalonica contained 11,620 COG categories, 9,827 GO terms, and 4,607 KEGG pathways through integration of data from InterProScan and eggNOG annotations. Moreover, visualizations of repeat elements, gene density, and GC content for each chromosome were generated using TBtools50.
Data Records
The transcriptome, Hi-C, Illumina, and ONT data can be accessed using the identification numbers SRR2930157051, SRR2930157152, SRR2930157253, and SRR2930157354, respectively. The genome assembly result is available in the NCBI database under the number GCA_040436485.155. We have deposited the genome annotation results in Figshare56.
Technical Validation
Two separate methods were employed to assess the quality of the assembled genome. Firstly, we evaluated assembly completeness utilizing BUSCO v5.0.457 against the Insecta reference gene set (n = 1,367). The assembly exhibited a BUSCO completeness of 99.0% BUSCO genes, comprising 98.6% single-copy, 0.4% duplicated, 0.3% fragmented, and 0.7% missing. Subsequently, we calculated the mapping rates of aligned reads from ONT, Illumina, and RNA data to the assembled genome, resulting in mapping rates of 99.22%, 95.47%, and 89.80%, respectively.
Code availability
The genome assembly and annotation scripts in this article were uploaded to figshare56. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
Ahmed, T. Z., Sarwar, M., Ijaz, M. & Sajjad, M. Biodiversity and Faunistic Studies of the Family Pyralidae (Lepidoptera) from Pothwar Region, Punjab, Pakistan. Pak. J. Life Soc. Sci. 15(2), 126–132 (2017).
Sabr, A. J. & Taha, Z. K. Review of family Pyralidae Latreille, 1809 (Lepidoptera: Pyraloidea). BioGecko. 12(03), 2230–5807 (2023).
Jin Roh, S. H., Park, S. H., Kim, S. Y., Kim, Y. S. & Choi, S. J. H. A new species of Galleria Fabricius (Lepidoptera, Pyralidae) from Korea based on molecular and morphological characters. ZooKeys. 970, 51–61 (2020).
Atwal, A. S. & Dhaliwal, G. S. Agricultural pests of South Asia and their management. Kalyani Publishers, New Delhi, India, (2008).
Samodra, H. & Ibrahim, Y. B. Effectiveness of selected entomopathogenic fungi in packed rice grain at room temperature against Corcyra cephalonica Stainton. Asean J. Sci. Technol. Develop. 23(3), 183–192 (2006).
Vincent, A., Singh, D. & Mathew, I. L. Corcyra cephalonica: A serious pest of stored products or a factitious host of biocontrol agents? J Stored Prod Res. 94, 101876 (2012).
Adom, M. et. Suitability of three Lepidopteran host species for mass-rearing the egg parasitoid Trichogrammatoidea eldanae Viggiani (Hymenoptera: Trichogrammatidae) for biological control of cereal stemborers. Int. J. Trop. Insect Sci. 41(1), 295–302 (2021).
Ghimire, M. N. & Phillips, T. W. Suitability of different lepidopteran host species for development of Bracon hebetor (Hymenoptera: Braconidae). Environ. Entomol. 39(2), 449–458 (2010).
Nathan, S. S., Kalaivani, K., Mankin, R. W. & Murugan, K. Effects of millet, wheat, rice, and sorghum diets on development of Corcyra cephalonica (Stainton) (Lepidoptera: Galleriidae) and its suitability as a host for Trichogramma chilonis Ishii (Hymenoptera: Trichogrammatidae). Environ. Entomol. 35(3), 784–788 (2006).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 11, 1432 (2020).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107 (2024).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Steinegger, M. & Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive datasets. Nat. Biotechnol. 35, 1026–1028 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Dudchenko, O. et al. Twelve years of SAMtools and BCFtools. GigaScience. 10(2), giab008 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna. 6, 11 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89 (2016).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating noncoding RNAs in complete genomes. Nucleic Acids Res 33, D121–124 (2005).
Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 491 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom. Bioinform. 3, lqaa108 (2021).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom. Bioinform. 2, lqaa26 (2020).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Kriventseva, E. V. et al. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics. 19, 189 (2018).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome research. 25, 445–458 (2015).
Yamamoto, K. & Shimada, T. High-quality genome assembly of the silkworm, Bombyx mori. Insect Biochem Mol Biol. 107, 53–62 (2019).
Lohse, K. et al. The genome sequence of the small white, Pieris rapae (Linnaeus, 1758). Wellcome Open Res. 6, 273 (2021).
Xiao, H. M. et al. Spodoptera frugiperda facilitated its rapid global dispersal and invasion. Mol Ecol Resour. 20(4), 1050–68 (2020).
Ma, W. et al. A chromosome-level genome assembly reveals the genetic basis of cold tolerance in a notorious rice insect pest, Chilo suppressalis. Mol Ecol Resour. 20(1), 268–282 (2020).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 46, D1282 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 13, 1194–1202 (2020).
NCBI Sequence Read Archive (Transcriptome data) https://identifiers.org/ncbi/insdc.sra:SRR29301570 (2024).
NCBI Sequence Read Archive (HI-C data) https://identifiers.org/ncbi/insdc.sra:SRR29301571 (2024).
NCBI Sequence Read Archive (Illumina short genome reads) https://identifiers.org/ncbi/insdc.sra:SRR29301572 (2024).
NCBI Sequence Read Archive (Nanopore long reads) https://identifiers.org/ncbi/insdc.sra:SRR29301573 (2024).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_040436485.1 (2024).
Jin, J. Genome annotation of Corcyra cephalonica. figshare Dataset. https://doi.org/10.6084/m9.figshare.25904035 (2024).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Acknowledgements
This study was supported by grants from the Youth Support Project of Jiangsu Vocational College of Agriculture and Forestry (2022kj27) to YHD.
Author information
Authors and Affiliations
Contributions
Z.Y. contributed to the research design. Y.D., J.J., Y.L. and M.F. collected the samples. Y.D., and J.J. analyzed the data. Y.D. and Z.Y. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, Y., Jin, J., Fang, M. et al. A chromosomal-level genome assembly of Corcyra cephalonica Stainton (Lepidoptera: Pyralidae). Sci Data 11, 1118 (2024). https://doi.org/10.1038/s41597-024-03967-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03967-w
