
Abstract
In this study, a chromosome-level genome of the tropical sea cucumber Stichopus monotuberculatus was generated by a combination of Nanopore long-read, Illumina short-read, and Hi-C sequencing technologies. The final assembly was 810.54 Mb in length, with contig N50 and scaffold N50 values of 10.15 Mb and 35.36 Mb, respectively. This assembly comprised 23 pseudo-chromosomes, covering 99.82% of the genome. Completeness analysis using BUSCO indicated that 97.8% of the metazoan conserved genes were presented in their entirety. A total of 29,596 protein-coding genes were predicted, with functional annotations available for 94.43% of these genes. The high-quality genome assembly produced in this study may provide an essential foundation for future researches on resource conservation and genetic breeding of S. monotuberculatus.
Similar content being viewed by others


Background & Summary
Sea cucumbers, belong to the phylum Echinodermata and the class Holothuroidea, are one of the largest groups of marine invertebrates found in diverse habitats on the sea floor worldwide1. The number of known sea cucumber species exceeds 1,800 in global2, of which over 80 species hold significant economic value, encompassing both dietary and medicinal applications3. In addition, sea cucumbers play crucial roles in the marine ecosystem through bioturbation, organic matter processing, nutrient recycling, seawater chemistry balancing, biodiversity supporting and energy transferring in the food chain1. It has been further reported that sea cucumbers safeguard coral reefs by mitigating disease4. However, the enormous commercial demand has led to overfishing, resulting in a significant decrease in many sea cucumbers’ populations5. Therefore, artificial breeding and release of seedlings were used to restore the wild sea cucumber resources6,7.
Stichopus monotuberculatus is a tropical sea cucumber that is commonly distributed in the coral reefs of the Indo-Pacific Ocean, ranging from the Red Sea and Madagascar to Easter Island, and from Japan to Australia (Fig. 1a)3,8. Characterized by the unique morphology, the adult S. monotuberculatus typically grows to ~20 cm in length, displaying a brownish-yellow body wall and a quadrangular shape (Fig. 1a)8,9. S. monotuberculatus is similar to Stichopus horrens and Stichopus naso in appearance, whereas genetic barcoding indicates that S. monotuberculatus and S. horrens fall within the same clade10,11. The abundant high-quality protein and collagen fibers in the body wall of S. monotuberculatus make it highly valuable for consumption12, earning it a premium edible sea cucumber in Asia3. In order to meet the commercial demand and restore the damage of wild resources due to overfishing, the artificial spawning of S. monotuberculatus has recently been developed9, and various related aquaculture techniques were also continually improved8,13,14. Moreover, research were further performed on taxonomic clarification10,15, sex determination16, genetic diversity17, food resource18, gut microbiota19 and immune mechanism20,21,22,23,24,25 in S. monotuberculatus. Despite the mitochondria DNA26, transcriptomic sequencing27, and preliminary unassembled genome draft28 of S. monotuberculatus having been conducted, there remains a significant gap in integrated genomic research, restricting a more comprehensive understanding of the evolutionary and genetic traits this species.

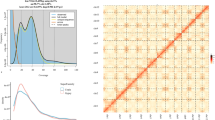
S. monotuberculatus and its genomics feature. (a) Appearance and global distribution of S. monotuberculatus; (b) 21-mer seq frequency distribution in the S. monotuberculatus genome; (c) Hi-C contact heatmap of the S. monotuberculatus genome.
In this study, we utilized a combination of Nanopore long-read sequencing, Illumina short-read sequencing, and Hi-C technology to produce a high-quality chromosome-level genome of S. monotuberculatus. The final assembly is estimated to be 810.54 Mb, with a contig N50 of 10.15 Mb and a scaffold N50 of 35.36 Mb, consisting of 23 pseudochromosomes and achieving 99.82% assembly coverage. The Benchmarking Universal Single-Copy Orthologs (BUSCO) integrity assessment indicates that 97.8% of the conserved metazoan genes are complete. The S. monotuberculatus genome contains 29,596 protein-coding genes, with 94.43% (27,948 genes) annotated with functional information. This high-quality genome assembly will facilitate a deeper understanding of the genomic structure and genetic traits of S. monotuberculatus, laying a solid foundation for future wild resource conservation, genetic breeding and aquaculture. Furthermore, this study can also give new insights to evolution and ecological adaptation mechanisms of echinoderms.
Methods
Sample collection and nucleic acid extraction
All samples used in this study were obtained from adult female S. monotuberculatus, collected from the natural habitat at Tanmen Port, Qionghai City, Hainan Province (19.33° N, 110.49° E). The longitudinal muscles of a female specimen were excised, washed three times with phosphate-buffered saline (PBS), quickly frozen in liquid nitrogen, and subsequently stored at −80 °C. High-quality DNA was extracted from the longitudinal muscles using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) to facilitate both long-read and short-read whole-genome sequencing.
Transcriptomic sequencing was carried out on 9 tissues, including the body wall, coelomocytes, intestine, muscle, oral tentacles, Polian vesicles, respiratory tree, rete mirabile and skin. The coelomocytes were harvested from coelomic fluids that were filtered by 100-μm sterile nylon mesh and centrifuged immediately at 4 °C and 1,000 × g for 10 min. Other tissues were carefully separated using scissors and forceps, then processed similarly to the muscle specimen. Total RNA was extracted using the RNAprep Pure Plant Plus Kit (Tiangen Biotech Co. Ltd., Beijing, China) for transcriptomic sequencing.
Library preparation and sequencing
For Illumina short-read sequencing, high-quality DNA was randomly fragmented using the Covaris ultrasonic disruptor (Woburn, MA, USA). The sequencing pair-end libraries, with a 350 bp insert size, were prepared using the Nextera DNA Flex Library Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed on the Illumina NovaSeq6000 platform. Low-quality reads were removed from raw reads through the SOAPnuke (v2.1.4)29 tool and clean data were used for subsequent analyses. A total of 148.04 Gb of short-read data (equivalent to a coverage of 182.64×) were obtained (Table 1).
For long-read Nanopore sequencing, libraries were prepared using the SQK-LSK110 ligation kit (Oxford Nanopore Technologies, Oxford, UK). The libraries were loaded onto primed R9.4 Spot-On Flow Cells and sequenced on a PromethION sequencer (Oxford Nanopore Technologies) with a 48-h run. Base-calling analysis of raw data was performed using the Oxford Nanopore GUPPY (v0.3.0)30. A total of 126.07 Gb of Nanopore continuous long-read data (equivalent to a coverage of 155.54×) were obtained (Table 1, Table 2).
For Hi-C sequencing with freshly harvested muscle samples, a formaldehyde cross-linking step was performed, followed by digestion of DpnII enzyme (NEB, Ipswich, MA, USA). In situ Hi-C chromosome conformation capture was performed following the DNase-based method31. The libraries were sequenced in 150 bp paired-end mode on an Illumina NovaSeq, resulting in 194.07 Gb of clean data (Table 1; Table 3).
For transcriptomic sequencing, 2 μg total RNA was used in a sample. Sequencing libraries were generated using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB), and index code was added for each sample. The sequencing was performed for 9 tissues, employing a strand-switching approach using Illumina HiSeq X Ten platform. A total of 174.53 Gb of clean data were generated in final.
Genome assessment and assembly
The K-mer analysis was conducted to estimate the genome size, proportion of repetitive sequences and heterozygosity using the Jellyfish (v2.3.0)30 and GenomeScope (v2.0.0)32. Jellyfish was used to count K-mer from the raw sequencing reads, configuring the K-mer and hash sizes at 21 M and 100 M, respectively (Fig. 1b; Table 4). Employing 10 threads, the analysis was accounted for both DNA strands. The K-mer histogram generated was further analyzed via GenomeScope, setting the K-mer size at 21 and assuming a diploidy level of 2. After discarding K-mers reflecting abnormal depth, the analysis yielded 120,544,041,989 K-mers, with a peak depth at 159 (Table 4). Based on these results, the S. monotuberculatus genome size was estimated to be 735.23 Mb, comprising about 1.86% heterozygosity and 29.67% repetitive sequences.
The initial long-read assembly of genome was performed using SMARTdenovo33, followed by polishing with nanopore sequencing data by Racon (v1.4.11)34. The subsequent error correction was accomplished by integrating 148.04 Gb of Illumina sequencing data via the Pilon (v1.23)35 software. Modifications to address heterozygosity were conducted using the Purge_haplotigs (v1.0.4)36 pipeline. Hi-C data was processed through the ALLHiC (v1.1)36 pipeline to organize scaffolds into chromosome-length sequences, supporting accurate scaffold placements into 23 pseudo-chromosomes (Fig. 1c; Table 3). The chromosomal architecture and diverse genomic features were visualized using Circos software (v0.69)37 (Fig. 2; Table 5). The assembly spans 810.55 Mb across 42 scaffolds, achieving a scaffold N50 of 35.36 Mb (Table 6).

Circos plot depicting the features of the S. monotuberculatus genome. From inner to outer circles: chromosomes, gene densities, repeat sequences, SNP percentage, and NGS sequencing depth. Average values for these features are 0.600, 0.200, 0.019 and 173, respectively. The windows size for all circles was 500k bp.
Repetitive element annotation
RepeatMasker38 (v 4.09 with RepBase 20181026) and EDTA39 (a whole-genome de-novo TE annotation pipeline) were used to create High-quality TE annotations. The EDTA pipeline included LTRharvest, the parallel version of LTR_FINDER, LTR_retriever, GRF, TIR-Learner, HelitronScanner, and RepeatModeler along with customized filtering scripts. The gene-like sequences and redundancy results were removed using nucleotide coding sequences (CDS). Overall, sequences constituting 25.02% of the assembled genome were identified as repeats, of which the most abundant repetitive element was DNA elements (20.3%), followed by long interspersed nuclear elements (LINEs, 3.24%), short interspersed nuclear elements (SINEs, 0.16%), SINEs (0.16%), terminal repeats (LTRs, 0.06%) (Fig. 3a; Table 7).

Prediction and annotation of repetitive element (a) and Noncoding RNA (b).
Noncoding RNA annotation
Ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs) were predicted by Barrnap (v0.9) and tRNAscan-SE (v2.0.11)40 using default parameters, respectively. For other non-coding RNAs, such as small nuclear RNAs (snRNAs) and microRNAs (miRNAs), an alignment with the Rfam database (v14.8)41 was conducted, followed by annotation using Infernal (v1.1.4)42. A total of 19 miRNAs, 2,023 tRNAs, 444 rRNAs, and 202 snRNAs were identified in the S. monotuberculatus genome (Fig. 3b; Table 8).
Gene prediction and functional annotation
To obtain a high-quality gene set derived from the genome, three methods, namely RNAseq, homology, and de novo, were employed, and the results were integrated using MAKER3 (v3.01.03)43. For RNAseq, a total of 28 samples from 9 different tissues were sequenced, resulting in the generation of 186.82 Gb of data (Table 9). The reads were aligned to the genome using HISAT2 (v2.2.1)44 and possible gene structures were assembled using StringTie (v2.1.7)45. For Homology-based predictions, protein sets selected from 6 representative genomes, including Homo sapiens (GCA_000001405.29), Drosophila melanogaster (GCA_000001215.4), Strongylocentrotus purpuratus (GCF_000002235.5)46, Acanthaster planci (GCF_001949145.1)47, A. japonicus (GCA_037975245.1)48, and Holothuria leucospilota (GCA_029531755.1)49, were aligned to the S. monotuberculatus genome with BLASTx50. For de novo method, genes were predicted using Augustus (v3.3.2)51 and Fgenesh, and integrated within MAKER3. Genes supported only by the de novo method were retained if they aligning to UniProt, as they may represent fast-evolving or novel genes in the species. A total of 29,596 genes were identified, with 79.4% supported by at least two methods, and 3,468 genes (12.5%) supported only by the de novo method. The comparison of the number and features of predicted genes in the S. monotuberculatus genome with those of 4 other echinoderm species including S. purpuratus46, A. planci47, A. japonicus48, and H. leucospilota49 were shown in Fig. 4a & Table 10.

Prediction and annotation of gene, and gene family analysis. (a) Prediction and annotation of gene; (b) Venn diagram of common and unique gene family in 4 sea cucumbers; (c) Phylogenetic and gene family evolution analysis among 9 echinoderm species. The scale below represents the divergence time. The number of expanded (+green) and contracted (−red) gene families were shown alongside the species.
Functional annotation of the predicted protein-coding genes was performed by BLASTp (v2.11.0, e-value 1e-5)52 against entries in the UniProt53 and UniProtKB/Swiss-Prot databases. Domain and GO information were identified using InterProScan (v5.52-86.0)54, resulting in successful annotation of 96.05% of the genes to the existing databases (Table 11). A common set of 8,844 genes is shared among all 4 species belonging to the class Holothuroidea, namely, A. japonicus48, S. monotuberculatus, H. leucospilota49, and H. scabra. The number of genes specific to each species are 893, 727, 1,380, and 688 in A. japonicus, S. monotuberculatus, H. leucospilota, and H. scabra, respectively. A Venn diagram is presented to visually illustrate both the shared characteristics and differences in the gene profiles of these 4 sea cucumber species (Fig. 4b).
Gene family analysis
Gene family clustering was conducted with 9 species belonging to the phylum Echinodermata, including A. planci (GCF_001949145.1), Lytechinus variegatus (GCA_018143015.1), S. purpuratus (GCF_000002235.5), Chiridota heheva (GCA_020152595.1), S. monotuberculatus (PRJNA1123322), A. japonicus (GCA_037975245.1), Holothuria glaberrima (GCA_009936505.2), Holothuria scabra (PRJNA1074116) and H. leucospilota (GCA_029531755.1), using OrthoFinder55 with MMseqs. 256, followed by phylogenetic tree construction based on single-copy genes. The analysis of gene families’ expansion and contraction was carried out across using CAFE (v2.1)57 (Fig. 4c). The number of gene families in the Most Recent Common Ancestor (MRCA) was determined to be 8915. Within the class Stichopodidae, there were 351 expanded gene families and 269 contracted gene families. Among them, 29 expansions and 22 contractions were found to be statistically significant (p < 0.01). In the case of S. monotuberculatus, there were 687 expanded gene families and 956 contracted gene families, with 68 expansions and 155 contractions exhibiting statistical significance (p < 0.01), possibly indicating adaptations to varying environmental conditions or ecological niches.
Data Records
All the sequencing reads and the chromosome-level genome assembly sequences related to this project have been deposited in NCBI BioProject PRJNA112332258. In this study, whole genome sequencing reads are available under the accession numbers SRR29673227 to SRR29654456, while RNA-seq sequencing reads can be found under the accession numbers SRR29673218 to SRR29673229. The whole genome shotgun project has been deposited at DDBJ/ENA/GenBank under the accession number JBEVTQ00000000059. Further related data, including assembly, annotation, functional annotation, and gene families, have been submitted to the Figshare database60.
Technical Validation
Nucleic acid quality
The DNA quality and concentration were assessed using 0.75% agarose gel electrophoresis, NanoDrop One spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). DNA samples showing slight degradation were deemed suitable for sequencing library preparation. RNA purity was evaluated with the kaiaoK5500® Spectrophotometer (Kaiao, Beijing, China). RNA integrity and concentration were determined using the RNA Nano 6000 Assay Kit on the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). Samples with an RNA integrity number (RIN) exceeding 9.50 were considered appropriate for library construction.
Genome assembly and annotation quality
The QV pipeline of Merqury61 was used to estimate the assembly QV based on k-mer analysis. The script “best_k.sh” in Merqury was employed to determine the optimal k-mer length, which was found to be 19. Meryl was then utilized to calculate the number of k-mers in the Illumina WGS reads with default settings. The QV evaluation was performed in Merqury using the output from Meryl and the assembly. The findings demonstrated a k-mer completeness of 79.10% and a k-mer-based QV of 43.36.
To assess the integrity and precision of the genome, alignment of sequenced reads was performed against the genome. High alignment integrity indicates robust genome integrity. Utilizing Illumina short-read sequencing, the genome was sequenced at the coverage of 182.64×, resulting in the generation of 148 Gb of high-quality reads. This approach achieved a mapping rate of 99.57%, with 90.43% of reads properly paired. The genome exhibited a coverage of 99.67%, with regions exceeding 30× coverage representing 99.01% of the genome (Table 12).
Genome completeness was further evaluated through BUSCO analysis using the metazoa_odb10 dataset, which contains 954 conserved genes across 65 genomes (Table 13). The analysis revealed an impressive overall completeness of 97.8%, with 94.3% of BUSCOs being complete, and minor proportions being fragmented (3.5%) or missing (2.2%). These results highlight the strength of our assembly and demonstrate the excellent quality of the genome. These metrics confirm the completeness and accuracy of our genome assembly, providing valuable data for further wild resource conservation, genetic breeding and aquaculture of S. monotuberculatus.
Code availability
All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software. The settings and parameters of software were listed below:SOAPnuke (v2.1.4): Used for filtering low-quality reads from Illumina short-read sequencing data with default parameters.GUPPY (v0.3.0): Used for base-calling analysis of Nanopore sequencing data with default parameters.Jellyfish (v2.3.0): Used for K-mer counting in genome size estimation, K-mer size is 19.GenomeScope (v2.0.0): Applied for analyzing K-mer histograms and estimating genome characteristics such as size, heterozygosity, and repetitive content with default parameters.SMARTdenovo: Used for initial assembly of long-read Nanopore data with default parameters.Racon (v1.4.11): Used for polishing the assembly with Nanopore data with default parameters.Pilon (v1.23): Applied for further error correction using Illumina data with default parameters.Purge_haplotigs (v1.0.4): Utilized for addressing heterozygosity in the genome assembly with -j 80 -s 80. ALLHiC (v1.1): Used for processing Hi-C data and organizing scaffolds into chromosome-length sequences. Merqury (v1.3): Used for genome assembly quality with best K-mer 19. BUSCO (v5.7.1): Applied for genome completeness assessment with metazoa_odb10 dataset.Circos (v0.69): Used for visualizing chromosomal architecture and various genomic features.RepeatMasker (v4.09): Applied for repetitive element annotation with default parameters.EDTA: Used for de novo transposable element (TE) annotation with default parameters.Barrnap (v0.9): Used for predicting ribosomal RNAs (rRNAs) with default parameters.tRNAscan-SE (v2.0.11): Used for transfer RNA (tRNA) prediction with default parameters.Infernal (v1.1.4): Used for annotating small nuclear RNAs (snRNAs) and microRNAs (miRNAs) with default parameters. MAKER3 (v3.01.03): Used for integrating gene prediction results with default parameters.HISAT2 (v2.2.1): Used for aligning RNA-seq reads to the genome with default parameters.StringTie (v2.1.7): Used for assembling possible gene structures from RNA-seq data with default parameters. Augustus (v3.3.2): Used for de novo gene prediction with general sets.OrthoFinder: Used for gene family clustering with mmseq2 aligner.MCMCTree from PAML (v4.10.7): Used for estimating divergence times.Evolview2: Used for visualizing phylogenetic trees.CAFE (v2.1): Used for gene family expansion and contraction analysis.
References
Purcell, S. W., Conand, C., Uthicke, S. & Byrne, M. Ecological roles of exploited sea cucumbers. Oceanogr Mar Biol 54, 367–386 (2016).
Ahyong, S. et al. World register of marine species (WoRMS). WoRMS Editorial Board (2024).
Purcell, S. W. et al. Commercially important sea cucumbers of the world – Second edition. Food and Agriculture Organization of the United Nations FAO (2023).
Clements, C. S., Pratte, Z. A., Stewart, F. J. & Hay, M. E. Removal of detritivore sea cucumbers from reefs increases coral disease. Nat Commun 15, 1338 (2024).
Anderson, S. C., Flemming, J. M., Watson, R. & Lotze, H. K. Serial exploitation of global sea cucumber fisheries. Fish Fish 12, 317–339 (2011).
Z, E. et al. Applications of Environmental DNA (eDNA) in Monitoring the endangered status and evaluating the stock enhancement effect of tropical sea cucumber Holothuria Scabra. Mar Biotechnol 25, 778–789 (2023).
Yang, Y. et al. Pipeline for identification of genome-wide microsatellite markers and its application in assessing the genetic diversity and structure of the tropical sea cucumber Holothuria leucospilota. Aquacult Rep 37, 102207 (2024).
Cheng, C. et al. Aquaculture of the tropical sea cucumber, Stichopus monotuberculatus: Induced spawning, detailed records of gonadal and embryonic development, and improvements in larval breeding by digestive enzyme supply in diet. Aquaculture 540, 736690 (2021).
Hu, C. et al. Larval development and juvenile growth of the sea cucumber Stichopus sp. (Curry fish). Aquaculture 300, 73–79 (2010).
Byrne, M., Rowe, F. & Uthicke, S. Molecular taxonomy, phylogeny and evolution in the family Stichopodidae (Aspidochirotida: Holothuroidea) based on COI and 16S mitochondrial DNA. Mol Phylogenet Evol 56, 1068–1081 (2010).
Uthicke, S., Byrne, M. & Conand, C. Genetic barcoding of commercial Beche-de-mer species (Echinodermata: Holothuroidea). Mol Biol Evol 10, 634–646 (2010).
Zhong, M., Chen, T., Hu, C. & Ren, C. Isolation and characterization of collagen from the body wall of sea cucumber Stichopus monotuberculatus. J Food Sci 80, C671–C679 (2015).
Gray, B. C. T., Byrne, M., Clements, M., Foo, S. A. & Purcell, S. W. Length-weight relationship for the dragonfish, Stichopus cf. monotuberculatus (Holothuroidea). Fish Res 268, 106851 (2023).
Huang, L. et al. Effects of acute salinity stress on physiology and immunoenzymatic activity in juvenile sea cucumber, Stichopus monotuberculatus. Aquaculture 578, 740094 (2024).
Wen, J. et al. The application of PCR–RFLP and FINS for species identification used in sea cucumbers (Aspidochirotida: Stichopodidae) products from the market. Food Control 21, 403–407 (2010).
Wu, F. et al. Identification of sex-specific molecular markers and development of PCR-based sex detection techniques in tropical sea cucumber (Stichopus monotuberculatus). Aquaculture 547, 737458 (2022).
Yuan, L., Hu, C., Zhang, L. & Xia, J. Population genetics of a tropical sea cucumber species (Stichopus monotuberculatus) in China. Conserv Genet 14, 1279–1284 (2013).
Jia, C. et al. Comparative analysis of in situ eukaryotic food sources in three tropical sea cucumber species by metabarcoding. Animals-Basel 12, 2303 (2022).
Jia, C. et al. Comparative Analysis of Gut Bacterial community composition in two tropical economic sea cucumbers under different seasons of artificial environment. Int J Mol Sci 25, 4573 (2024).
Yan, A. et al. Identification and functional characterization of a novel antistasin/WAP-like serine protease inhibitor from the tropical sea cucumber, Stichopus monotuberculatus. Fish Shellfish Immun 59, 203–212 (2016).
Ren, C. et al. The first echinoderm poly-U-binding factor 60 kDa (PUF60) from sea cucumber (Stichopus monotuberculatus): Molecular characterization, inducible expression and involvement of apoptosis. Fish Shellfish Immun 47, 196–204 (2015).
Ren, C., Chen, T., Jiang, X., Wang, Y. & Hu, C. The first characterization of gene structure and biological function for echinoderm translationally controlled tumor protein (TCTP). Fish Shellfish Immun 41, 137–146 (2014).
Ren, C. et al. The first echinoderm gamma-interferon-inducible lysosomal thiol reductase (GILT) identified from sea cucumber (Stichopus monotuberculatus). Fish Shellfish Immun 42, 41–49 (2015).
Chen, T. et al. Calmodulin of the tropical sea cucumber: Gene structure, inducible expression and contribution to nitric oxide production and pathogen clearance during immune response. Fish Shellfish Immun 45, 231–238 (2015).
Ren, C. et al. Two proprotein convertase subtilisin/kexin type 9 (PCSK9) paralogs from the tropical sea cucumber (Stichopus monotuberculatus): Molecular characterization and inducible expression with immune challenge. Fish Shellfish Immun 56, 255–262 (2016).
Zhong, S., Liu, Y., Zhao, Y. & Huang, G. The complete mitochondrial genome of sea cucumber Stichopus monotuberculatus (aspidochirotida: Stichopodidae). Mitochondrial Dna B 4, 3305–3306 (2019).
Ma, B. et al. Integrative Application of transcriptomics and metabolomics provides insights into unsynchronized growth in sea cucumber (Stichopus monotuberculatus). Int J Mol Sci 23, 15478 (2022).
Zhong, S. P. et al. The draft genome of the tropical sea cucumber Stichopus monotuberculatus (Echinodermata, Stichopodidae) reveals critical genes in fucosylated chondroitin sulfates biosynthetic pathway. Front Genet 14, 1182002 (2023).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2017).
Wick, R. R., Judd, L. M. & Holt, K. E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol 20, 129 (2019).
Ramani, V. et al. Sci-Hi-C: A single-cell Hi-C method for mapping 3D genome organization in large number of single cells. Methods 170, 61–68 (2020).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Liu, H., Wu, S., Li, A. & Ruan, J. SMARTdenovo: a de novo assembler using long noisy reads. GigaByte 2021, 15–15 (2021).
Vaser, R., Sovic, I., Nagarajan, N. & Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746 (2017).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plos One 9, e112963 (2014).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res 19, 1639–1645 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. P Natl Acad Sci USA 117, 9451–9457 (2020).
Ou, S. J. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20, 275 (2019).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–D124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18, 188–196 (2008).
Kim, D., Landmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Elsik, C. G. et al. The genome of the sea urchin. Science 315, 766–766 (2007).
Hall, M. R. et al. The crown-of-thorns starfish genome as a guide for biocontrol of this coral reef pest. Nature 544, 231–234 (2017).
Sun, L. A., Jiang, C. X., Su, F., Cui, W. & Yang, H. S. Chromosome-level genome assembly of the sea cucumber Apostichopus japonicus. Sci Data 10, 454 (2023).
Chen, T. et al. The Holothuria leucospilota genome elucidates sacrificial organ expulsion and bioadhesive trap enriched with amyloid-patterned proteins. P Natl Acad Sci USA 120, e2213512120 (2023).
Camacho, C. et al. BLAST plus: architecture and applications. Bmc Bioinformatics 10, 421 (2009).
Gao, Z. et al. TSTD:A Cross-modal Two Stages Network with New Trans-decoder for Point Cloud Semantic Segmentation. In: Pattern Recognition and Computer Vision, PRCV 2023, Part VIII (2024).
Li, Y. C. & Lu, Y. C. BLASTP-ACC: Parallel Architecture and Hardware Accelerator Design for BLAST-Based Protein Sequence Alignment. IEEE T Biomed Circ S 13, 1771–1782 (2019).
UniProt C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480-D489.
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238 (2019).
Mirdita, M., Steinegger, M., Breitwieser, F., Söding, J. & Levy Karin, E. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 37, 3029–3031 (2021).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2021).
Chen, T. Stichopus monotuberculatus isolate:south_sea_xycz | cultivar:nanhai. NCBI BioProject https://identifiers.org/ncbi/insdc.sra:SRP517039 (2024).
Chen, T. Stichopus monotuberculatus isolate:south_sea_xycz | cultivar:nanhai Genome sequencing and assembly. GenBank https://identifiers.org/ncbi/insdc:JBEVTQ000000000 (2024).
Chen, T. Chromosome-level genome assembly and annotation of the tropical sea cucumber Stichopus monotuberculatus. Figshare https://doi.org/10.6084/m9.figshare.26124061 (2024).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Acknowledgements
This study was graciously supported by grants from the National Natural Science Foundation of China (42176132), the Guangxi Natural Science Foundation (2021GXNSFAA196074), the Science and Technology Program of Nansha District (NSJL202103), the Research on breeding technology of candidate species for Guangdong modern marine ranching (2024-MRB-00-001), the National Key R & D Program of China (2022YFD2401301), the Guangdong Province Project (2024A1515011418, 2024A1515010899), and the Innovation Team Project of High Level Local Universities from Shanghai Education Committee (HJWK-2021-21).
Author information
Authors and Affiliations
Contributions
Ting Chen, Chunhua Ren, Aifen Yan and Chaoqun Hu conceived and designed this study. Ting Chen, Yun Yang, Zhenyu Xie, Hao Long, Wenjie Pan, Zixuan E, Jiasheng Huang, Qianying Huang, Jianfeng Xu, Zepeng Zhang, Chuhang Cheng and Suzhong Yu collected the samples. Zhou Qin and Dingding Fan assembled and annotated the genome. Ting Chen, Yun Yang, Xuan Wang, Zhou Qin and Dingding Fan performed gene family and genome evolutionary analyses. Ting Chen, Yun Yang, Xuan Wang, Zhou Qin and Dingding Fan performed bioinformatic analyses. Ting Chen, Zhenyu Xie, Chunhua Ren, Hongyan Sun, Peng Luo, Xiao Jiang, Chang Chen, Yanhong Wang, Fajun Jiang, Aifen Yan, and Chaoqun Hu contributed reagents/analytic tools. Ting Chen, Yun Yang and Dingding Fan wrote the manuscript. Chunhua Ren, Aifen Yan and Chaoqun Hu revised it. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, T., Yang, Y., Wang, X. et al. Chromosome-level genome assembly and annotation of the tropical sea cucumber Stichopus monotuberculatus. Sci Data 11, 1245 (2024). https://doi.org/10.1038/s41597-024-03985-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03985-8
This article is cited by
-
A chromosomal-level genome assembly of the tropical sea cucumber Stichopus fusiformiossa
Scientific Data (2025)
-
Chromosome-level genome assembly of the sea cucumber, Colochirus anceps
Scientific Data (2025)
-
A chromosome-level genome assembly for the brown sea cucumber from the Galápagos Islands, Isostichopus fuscus
Scientific Data (2025)
-
Chromosome-level genome assembly and annotation of the tropical sea cucumber Bohadschia ocellate
Scientific Data (2025)
