
Abstract
The Arctic Ocean is experiencing significant global warming, leading to reduced sea-ice cover, submarine permafrost thawing, and increased river discharge. The East Siberian Sea (ESS) undergoes more significant terrestrial inflow from coastal erosion and river runoff than other Arctic seas. Despite extensive research on environmental changes, microbial communities and their functions in the ESS, which are closely related to environmental conditions, remain largely unexplored. Here, we investigated microbial communities in ESS surface sediments spanning latitudes from 73°N to 77°N using 16S rRNA amplicon sequencing, and reconstructed 211 metagenome-assembled genomes (MAGs) using shotgun metagenome sequencing. Taxonomic analysis identified 209 bacterial MAGs, with the predominant phyla Pseudomonadota (n = 82), Actinobacteriota (n = 38), Desulfobacterota (n = 23), along with 2 archaeal MAGs of Thermoproteota. Notably, 86% of the MAGs (n = 183) could not be classified into known species, indicating the potential presence of novel and unidentified microorganisms in the ESS. This dataset provides invaluable information on the microbial diversity and ecological functions in the rapidly changing ESS.
Similar content being viewed by others

Background & Summary
The Arctic is undergoing the most rapid climate changes, warming by 1.9 °C over the last 30 years which is two to three times faster than the global average1,2,3. As a consequence, the Arctic marine ecosystem is rapidly changing with reductions in sea-ice cover and submarine permafrost thawing3,4,5,6. Environmental changes in the coastal marine ecosystem are also accelerated by the terrestrial impacts. For example, freshwater and sedimentary deposits from enhanced terrestrial runoff pose a threat to shallow estuaries and coastal benthic ecosystems of the continental shelf6,7,8.
Marine sediments are the largest organic carbon reservoirs, supporting rich and diverse benthic microbial communities9. Benthic microorganisms play key roles in biochemical cycles through diverse metabolisms, including the oxidation of organic matter, the production of carbon oxide and other hydrocarbons, and the removal of sulfates9,10,11. The structure and function of benthic microbial communities are influenced by various environmental factors12,13,14,15,16,17,18,19. Previous studies on the benthic microbial communities in the coastal areas in the Beaufort Sea, Greenland, and Svalbard in the Arctic have revealed a close relationship between these communities and environmental variables including organic matter or salinity, which are largely linked to terrestrial impacts16,17,18,19. Since changes in organic matter and salinity in coastal environments affect microbial communities and their metabolisms, which play an increasingly significant role in coastal biogeochemistry and carbon fluxes, it is important to characterize microbial communities in the Arctic seas across a wide range of continental shelves. This characterization is crucial for understanding and predicting the responses and functional changes of microbial communities along the marine environmental gradient20.
The East Siberian Sea (ESS) is the widest of the Arctic Ocean shelf seas and the shallowest, with a mean depth of 52 m21. It is mostly underlain by subsea permafrost and is experiencing rapid warming22. With a vast coastline and significant sea-ice coverage, the ESS receives substantial terrestrial inflows from coastal erosion and river runoff, accounting for approximately 2.2 million tons and 1.9 million tons of organic carbon, respectively22. Additionally, emissions of dissolved organic matter from the subsea permafrost, which contain substantial amounts of organic carbon (ranging from 943 to 2,240 g C m−2 yr−1 at the continuous-discontinuous transition zone of subsea permafrost and from 10 to 55 g C m−2 yr−1 in the remaining shelf and slope sites) underscore the crucial role of the ESS in Arctic climate dynamics22. These processes also drive shifts in benthic microbial communities19. Changes in organic matter inputs have led to the dominance of specific bacterial groups, affecting the decomposition and remineralization of organic carbon23. Furthermore, alterations in the structure of microbial communities impact their function in terms of nutrient cycling processes24. However, studies on microbial structures and functions, which would provide important insights into microbial contributions to nutrient cycling in the ESS, have not been performed yet.
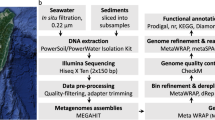
Here, we present 16S rRNA gene amplicon and shotgun metagenome sequencing datasets from surface sediments from 13 and 7 sites, respectively, in the ESS, covering latitudes from 73°N to 77°N (Fig. 1a and Tables 1–3). A schematic representation of the metagenomic analysis in this study is shown in Fig. 1b. Taxonomic classification by 16S rRNA amplicon sequencing revealed that the bacterial community was dominated by the phyla Pseudomonadota (51.1 ± 6.6%) followed by Bacteroidota (16.4 ± 9.5%), Planctomycetota (8.9 ± 3.8%), Acidobacteriota (6.5 ± 4.2%), and Actinomycetota (2.6 ± 2.0%) (Fig. 2a). In the archaeal community, Thaumarchaeota (70.9 ± 11.1%) and Euryarchaeota (27.7 ± 11.4%) were predominant (Fig. 2b). Some microbial taxa showed significant changes along the latitude, with the proportion of Alphaproteobacteria and Acidobacteriota increasing, while those of Bacteroidota, Deltaproteobacteria, and Thaumarchaeota decreased (Fig. 2a,b). At the amplicon sequence variant (ASV) level, bacterial and archaeal communities exhibited distinct community patterns according to different water depth (100 m), with significant analysis of similarities (ANOSIM) R values of 0.83 and 0.75 (p < 0.0001), respectively (Fig. 2c,d).

Sampling location and sequencing approaches. (a) Geographic location of samples of marine sediments. The exclusive economic zone (EEZ) is labeled as a gray dashed line. (b) Schematic representation of 16S rRNA gene amplicon and shotgun metagenome sequencing analysis pipeline.

The relative abundance of microbial communities at the phylum and class level for Pseudomonadota. (a) Bacterial community at the phylum and proteobacterial class level. Phyla classified as ‘Others’, which each have an average relative abundance of less than 0.5%, including 66 phyla such as Rhodothermota, Calditrichota, candidate division GN04, Candidatus phylum TM6, Spirochaetota, and Lentisphaerota, all of which have an average relative abundance of more than 0.1%. (b) Archaeal community at the phylum level. (c) Non-metric multidimensional scaling (NMDS) analysis for the bacterial community at the amplicon sequence variant (ASV) level. (d) NMDS analysis for the archaeal community at the ASV level. n/a, not applicable indicates that communities were not analyzed due to the low number of sequences obtained from each sample.
For metagenome sequencing, surface sediments at a depth of 1 centimeter below the seafloor (cmbsf) from 7 sites were used (Fig. 1; Table 1). The shotgun metagenome sequencing generated a total of 229.1 Gbp, with 31.9–34.0 Gbp per sample, and 1.51 billion paired-end reads, with an average of 216.8 million reads per sample (Table 3). After quality control to discard low-quality reads, 1.45 billion paired-end reads were retained, accounting for an average of 95.75% of raw reads. Subsequently, these metagenomic data were individually assembled into contigs. Using the metaWRAP (v1.3.2) pipeline25, we reconstructed 211 metagenome-assembled genomes (MAGs), and their quality metrics are summarized in Table S1 (see supplementary xlsx file). All the MAGs had >70% completeness and <10% contamination, and 61 high-quality MAGs (>90% completeness and <5% contamination) were obtained (Fig. 3a). Among high-quality MAGs, 28 MAGs showed >95% completeness and <5% contamination, and 3 MAGs represented >97% completeness and <1% contamination. The relationship between completeness and contamination represented a negative correlation (R = −0.17, p = 0.016; Fig. 3a). The genome size of the MAGs ranged from 1.11 to 5.93 Mbp, with an average of 2.67 Mbp. The majority of the genomes fell within 2 to 3 Mbp range (Fig. 3b; Table S2, see supplementary xlsx file). The 124 (58%) MAGs showed an N50 metric greater than 10 Kbp, with the longest value of 131 Kbp (Fig. 3c). The relationship between the genome size and N50 metric exhibited a positive correlation (R = 0.25, p = 0.00029; Fig. 3b). Half of the MAGs consisted of fewer than 300 contigs (Fig. 3d). The GC content of the MAGs ranged from 30.88% to 70.26% with an average of 54.5% (Table S1, see supplementary xlsx file).

Overview of 211 MAGs recovered from the East Siberian Sea. (a) The relationship between the completeness and contamination of MAGs. (b) The bar plot compares the genome sizes of MAGs. (c) The relationship between the genome size and N50 length of MAGs. (d) The bar plot compares the number of contigs of MAGs.
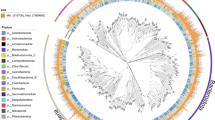
Using the Genome Taxonomy Database Toolkit (GTDB-tk, v.2.1.1)26, the taxonomic classification of the MAGs identified 209 bacteria and 2 archaea (Fig. 4; Table S2, see supplementary xlsx file). The two archaeal MAGs belonged to the family Nitropumilaceae of the phylum Thermoproteota. Among the 209 bacterial MAGs, 15 phyla were identified, with the most abundant being Pseudomonadota (n = 82), Actinobacteriota (n = 38), and Desulfobacterota (n = 23) (Figs. 4, 5). Notably, 86% of MAGs (n = 183) could not be taxonomically assigned to any entry in the Genome Taxonomy Database (GTDB), suggesting that most of these MAGs belonged to unknown microbial taxa at different taxonomical level including 2 orders, 9 families, 51 genera, and 121 species (2 archaea and 181 bacteria) (Table S2, see supplementary xlsx file). Based on an average nucleotide identity (ANI) > 95%27, 130 bacterial MAGs were classified into 48 species, with varying recovery across the sites while 81 MAGs were reconstructed from a single metagenome (Table S3, see supplementary xlsx file).

Taxonomic classification of 211 MAGs recovered from the East Siberian Sea. (a) The Sankey diagram represents the classification of MAGs at different taxonomic ranks using GTDB-tk26. Unclassified MAGs are not shown. (b) The bar plot indicates the taxonomic novelty of the constructed MAGs. The number of classified taxa at each taxonomic level corresponds to the summed number of taxa shown in Fig. 4a.

Phylogenetic tree of 209 bacterial MAGs constructed from 120 bacterial single-copy genes. The circle colors at the ends of the branches indicate the phylum of the corresponding MAG.
Our study unveiled the microbial communities and microbial genomes harbored in the sediments of the ESS. Microbial communities were clearly differentiated by water depth, which may be partially related to the impact of terrestrial input. In addition, the MAGs reconstructed from metagenome sequences revealed a high proportion of unknown genomes. These findings suggest that microbial communities in the ESS surface sediments are correlated with water depth and latitude, and that the benthic communities harbor a largely unexplored microbial diversity. To the best of our knowledge, this is the first report to recover microbial genomes from the surface sediments of the ESS. The presented datasets can be further used to understand the structure and function of microorganisms in the rapidly changing oceanic environment in the Arctic.
Methods
Sample preparation and sequencing of 16S rRNA gene amplicon and metagenome
Marine sediment samples were collected at 13 stations outside of the Russia’s exclusive economic zone (EEZ) of the ESS (Fig. 1; Table 1). Sampling was conducted using a box corer or multi corer in September of 2016 and 2019 during ARA07C and ARA10C cruises of the Korean ice-breaker RV Araon. The total length of the cores ranged from 27 to 65 cmbsf. Upon recovery, core sediments were sliced into 1 cm section (2–5 cm intervals for samples of ST16). The edges of each slice were removed, a portion of each 1 cm slice was subsampled for 16S rRNA gene sequences and metagenome analyses on board, and stored at –80 °C until analysis.
Genomic DNAs from core samples at depths of 1, 2, and 3 cmbsf at each station were extracted using the FastDNA spin kit for soil (MP Biomedicals, USA). DNA samples were submitted for PCR amplification, library preparation, and paired-end Illumina MiSeq sequencing (2×300 bp) to the Integrated Microbiome Resource (IMR), Dalhousie University, Canada (http://cgeb-imr.ca). Two primer sets were used independently to amplify bacterial and archaeal 16S rRNA genes. The primer pair 515 F (5′-GTGYCAGCMGCCGCGGTAA)/926 R (5′-CCGYCAATTYMTTTRAGTTT) was used to amplify bacterial 16S rRNA genes targeting V4-V5 regions, and the primer pair 956 F (5′-TYAATYGGANTCAACRCC)/1401 R (5′-CRGTGWGTRCAAGGRGCA) was utilized to amplify archaeal 16S rRNA genes targeting V6-V8 regions28,29. Amplicon sequencing was conducted using the paired-end (2 × 300 bp) Illumina MiSeq system (Illumina, USA) at IMR (Table 2). In total, 59 sequencing datasets were used for community analyses including 22 datasets obtained using the 956 F/1401 R primer set for archaeal community analysis, and 37 datasets using the 515 F/926 R primer set for bacterial community analysis (Tables 1, 2). Based on community similarity analysis of the 16S rRNA gene amplicon sequences from 13 sites at a depth of 1–3 cmbsf, 7 sites (ST07, ST05, ST04, ST03, ST02, ST08, and ST20) were selected and metagenome sequencing of sediments at 1 cmbsf was performed (Table 3). All metagenomic libraries were shotgun sequenced to generate 151 bp paired-end reads using the Illumina HiSeq X system (Illumina, USA) at Phyzen (Seongnam, Korea).
16S rRNA gene sequences processing
The adapter and primer sequences were removed using Cutadapt (v2.10)30 and the resultant sequences were processed using DADA2 (v0.9.5)31 to infer amplicon sequence variants (ASVs). For the quality trimming process, we applied the filtering option as maxN = 0, maxEE = c(2,2), and truncQ = 2. The low-quality tails of both reads were removed with truncLen = c(270,210). Denoising was performed after trimming based on the DADA2 error model. Sequences were dereplicated and a core sample inference algorithm was applied to the dereplicated data. Paired reads were merged together and chimeric sequences were removed. Following processes were performed after constructing a sequence table of ASVs to assign taxonomy using the mothur package (v1.44.1)32. Taxonomic assignments of representative ASV sequences were determined against the EzBiocloud database by sequence similarity searches33. After taxonomic assignment, archaea and unknown ASVs were removed for bacterial analysis, and bacteria and unknown ASVs were removed for archaeal analysis. Non-metric multidimensional scaling (NMDS) analysis was performed based on the Bray-Curtis dissimilarities with relative abundance matrix using the vegan package (v2.64) in R. An analysis of similarities (ANOSIM) was performed with 9,999 permutations using the vegan package (v2.64) in R.
Metagenomic assembly, binning, and refinement
Raw reads were introduced into Sickle (v1.33) (https://github.com/najoshi/sickle) to perform the quality control with the options of -n -q 20 -l 60. Filtered reads were then individually assembled by MEGAHIT (v1.2.9)34 using the option of --min-contig-len 500.
Assembled contigs with a length >1,000 bp were binned to recover MAGs using the metaWRAP (v1.3.2)25. This process employed tetranucleotide frequencies, GC content, and coverage as criteria, and included binning tools MaxBin2 (v2.2.6)35, MetaBAT2 (v2.12.1)36, and CONCOCT (v1.0.0)37 integrated within the metaWRAP pipeline. Afterward, the “bin_refinement” module in the metaWRAP was performed to improve the bin quality with options of -c 70 and -x 10 (completeness >70% and contamination <10%). The completeness and contamination of the bins were assessed using CheckM (v1.2.2)38 as part of the metaWRAP pipeline. The bins were then reassembled using the “reassemble_bins” module in the metaWRAP with options -c 70 and -x 10. To dereplicate multiple bin sets recovered from seven individual assemblies, dRep (v3.5)39 was used with a 95% ANI threshold. Finally, a total of 211 MAGs were retained (Table S2, see supplementary xlsx file).
Taxonomic assignment and phylogenetic assessment
Taxonomic classification of MAGs was performed using the “classify_wf” module in the Genome Taxonomy Database toolkit (GTDB-tk, v2.1.1)26 with the Genome Taxonomy Database (GTDB, Release 207 v2).
The phylogenetic tree of 209 bacterial MAGs was constructed with 120 bacterial single-copy marker genes which were obtained from the result of GTDB-tk analysis (Table S4, see supplementary xlsx file). Concatenated single-copy genes were aligned using MUSCLE (v3.8.31)40 with the default option, and the evolutionary distance between 209 MAGs was calculated using the maximum-likelihood method with bootstrap analyses of 1,000 replications on the MEGA 6.0 program (v6.06)41. The final tree was visually annotated using the Interactive Tree of Life (iTOL, v6)42.
Data Records
The 16S rRNA gene sequencing data, metagenome sequencing data, and reconstructed MAGs generated in this study are publicly available at the European Nucleotide Archive (ENA) under the accession number PRJEB7667243. The 211 reconstructed MAGs have been deposited at the DDBJ/ENA/GenBank database under accession numbers listed in Table S2 (see supplementary xlsx file).
Technical Validation
All software and parameters used in this study were described in the Methods section. The removal of adapter sequences was performed using Cutadapt, and low-quality reads were trimmed using Sickle. CheckM (v1.2.2)38 was used to assess the completeness and contamination of constructed MAGs. To investigate the distribution of MAGs between different 7 samples, all-against-all comparisons were performed using OrthoANI (v.140)44.
Usage Notes
This study provides 16S rRNA gene and shotgun metagenome sequencing datasets of surface sediments collected from the ESS, spanning latitudes from 73°N to 77°N. The comprehensive dataset from the ESS, where microbial communities have not been investigated, serves as a reference for comparing with other microbial communities and understanding their role in the rapidly changing Arctic.
Researchers should consider the sampling depth of surface sediments in this study. For 16S rRNA gene sequencing, sediments from depths of 1–3 cmbsf were sequenced, while for shotgun metagenome sequencing, only sediments from 1 cmbsf were used. Any interpretation of the data should take these depths into account. In addition, a total of 211 MAGs were categorized into 129 species based on all-against-all comparisons with an ANI >95%.
In the Methods section, we described the procedures for sampling, DNA extraction, library preparation, sequencing, and data processing and analysis used in this study. Detailed information about the samples is provided in Tables 1–3. Statistics for the constructed MAGs are listed in Tables S1, S2 (see supplementary xlsx file).
Code availability
Custom-designed scripts were not used to generate or process this dataset. The method section described and referenced all software versions and non-default parameters used in this study.
References
Grosse, G., Goetz, S., McGuire, A. D., Romanovsky, V. E. & Schuur, E. A. G. Changing permafrost in a warming world and feedbacks to the earth system. Environ Res Lett 11, https://doi.org/10.1088/1748-9326/11/4/040201 (2016).
Lenton, T. M. Arctic climate tipping points. Ambio 41, 10–22, https://doi.org/10.1007/s13280-011-0221-x (2012).
Walsh, J. E., Overland, J. E., Groisman, P. Y. & Rudolf, B. Ongoing climate change in the Arctic. Ambio 40, 6–16, https://doi.org/10.1007/s13280-011-0211-z (2011).
Carmack, E. C. et al. Freshwater and its role in the Arctic marine system: sources, disposition, storage, export, and physical and biogeochemical consequences in the Arctic and global oceans. J Geophys Res-Biogeo 121, 675–717, https://doi.org/10.1002/2015jg003140 (2016).
James, R. H. et al. Effects of climate change on methane emissions from seafloor sediments in the Arctic Ocean: A review. Limnol Oceanogr 61, S283–S299, https://doi.org/10.1002/lno.10307 (2016).
Peterson, B. J. et al. Increasing river discharge to the Arctic Ocean. Science 298, 2171–2173, https://doi.org/10.1126/science.1077445 (2002).
Holmes, R. M. et al. Seasonal and annual fluxes of nutrients and organic matter from large rivers to the Arctic Ocean and surrounding seas. Estuar Coast 35, 369–382, https://doi.org/10.1007/s12237-011-9386-6 (2012).
McGovern, M., Poste, A. E., Oug, E., Renaud, P. E. & Trannum, H. C. Riverine impacts on benthic biodiversity and functional traits: a comparison of two sub-Arctic fjords. Estuar Coast Shelf S 240, https://doi.org/10.1016/j.ecss.2020.106774 (2020).
Kallmeyer, J., Pockalny, R., Adhikari, R. R., Smith, D. C. & D’Hondt, S. Global distribution of microbial abundance and biomass in subseafloor sediment. P Natl Acad Sci USA 109, 16213–16216, https://doi.org/10.1073/pnas.1203849109 (2012).
Glöckner, F. O., Gasol, J. M., McDonough, N. & Calewaert, J.-B. Marine microbial diversity and its role in ecosystem functioning and environmental change. (European Science Foundation, 2012).
Whitman, W. B., Coleman, D. C. & Wiebe, W. J. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95, 6578–6583, https://doi.org/10.1073/pnas.95.12.6578 (1998).
Zhang, C. et al. Spatial patterns and co-occurrence networks of microbial communities related to environmental heterogeneity in deep-sea surface sediments around Yap Trench, Western Pacific Ocean. Sci Total Environ 759, 143799, https://doi.org/10.1016/j.scitotenv.2020.143799 (2021).
Balmonte, J. P., Teske, A. & Arnosti, C. Structure and function of high Arctic pelagic, particle-associated and benthic bacterial communities. Environmental Microbiology 20, 2941–2954, https://doi.org/10.1111/1462-2920.14304 (2018).
Bonthond, G. et al. Benthic microbial biogeographic trends in the North Sea are shaped by an interplay of environmental drivers and bottom trawling effort. ISME Commun 3, 132, https://doi.org/10.1038/s43705-023-00336-3 (2023).
Currie, A. A. et al. Sea Ice Dynamics Drive Benthic Microbial Communities in McMurdo Sound, Antarctica. Front Microbiol 12, 745915, https://doi.org/10.3389/fmicb.2021.745915 (2021).
Delpech, L. M. et al. Terrestrial inputs shape coastal bacterial and archaeal communities in a high Arctic fjord (Isfjorden, Svalbard). Front Microbiol 12, 614634, https://doi.org/10.3389/fmicb.2021.614634 (2021).
Garneau, M. É., Vincent, W. F., Terrado, R. & Lovejoy, C. Importance of particle-associated bacterial heterotrophy in a coastal Arctic ecosystem. J Marine Syst 75, 185–197, https://doi.org/10.1016/j.jmarsys.2008.09.002 (2009).
Hauptmann, A. L. et al. Upstream freshwater and terrestrial sources are differentially reflected in the bacterial community structure along a small Arctic river and its estuary. Front Microbiol 7, 1474, https://doi.org/10.3389/fmicb.2016.01474 (2016).
Walker, A. M., Leigh, M. B. & Mincks, S. L. Patterns in benthic microbial community structure across environmental gradients in the Beaufort Sea shelf and slope. Front Microbiol 12, 581124, https://doi.org/10.3389/fmicb.2021.581124 (2021).
McClelland, J. W., Holmes, R. M., Dunton, K. H. & Macdonald, R. W. The Arctic Ocean estuary. Estuar Coast 35, 353–368, https://doi.org/10.1007/s12237-010-9357-3 (2012).
Anderson, L. G. et al. East Siberian Sea, an Arctic region of very high biogeochemical activity. Biogeosciences 8, 1745–1754, https://doi.org/10.5194/bg-8-1745-2011 (2011).
Chen, M. L. et al. Subsea permafrost as a potential major source of dissolved organic matter to the East Siberian Arctic Shelf. Sci Total Environ 777, https://doi.org/10.1016/j.scitotenv.2021.146100 (2021).
Hoffmann, K., Hassenruck, C., Salman-Carvalho, V., Holtappels, M. & Bienhold, C. Response of bacterial communities to different detritus compositions in Arctic deep-sea sediments. Front Microbiol 8, 266, https://doi.org/10.3389/fmicb.2017.00266 (2017).
Blaud, A., Lerch, T. Z., Phoenix, G. K. & Osborn, A. M. Arctic soil microbial diversity in a changing world. Res Microbiol 166, 796–813, https://doi.org/10.1016/j.resmic.2015.07.013 (2015).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158, https://doi.org/10.1186/s40168-018-0541-1 (2018).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927, https://doi.org/10.1093/bioinformatics/btz848 (2019).
Richter, M. et al. Shifting the genomic gold standard for the prokaryotic species definition. PNAS. (2009).
Comeau, A. M., Li, W. K. W., Tremblay, J. E., Carmack, E. C. & Lovejoy, C. Arctic Ocean microbial community structure before and after the 2007 record sea ice minimum. Plos One 6, https://doi.org/10.1371/journal.pone.0027492 (2011).
Walters, W. et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, https://doi.org/10.1128/mSystems.00009-15 (2016).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12, https://doi.org/10.14806/ej.17.1.200 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583, https://doi.org/10.1038/nmeth.3869 (2016).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541, https://doi.org/10.1128/AEM.01541-09 (2009).
Yoon, S. H. et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67, 1613–1617, https://doi.org/10.1099/ijsem.0.001755 (2017).
Li, D. et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11, https://doi.org/10.1016/j.ymeth.2016.02.020 (2016).
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607, https://doi.org/10.1093/bioinformatics/btv638 (2016).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359, https://doi.org/10.7717/peerj.7359 (2019).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat Methods 11, 1144–1146, https://doi.org/10.1038/nmeth.3103 (2014).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055, https://doi.org/10.1101/gr.186072.114 (2015).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11, 2864–2868, https://doi.org/10.1038/ismej.2017.126 (2017).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–2729, https://doi.org/10.1093/molbev/mst197 (2013).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res https://doi.org/10.1093/nar/gkae268 (2024).
NCBI Bioproject. https://identifiers.org/ncbi/bioproject:PRJEB76672 (2024).
Lee, I., Ouk Kim, Y., Park, S. C. & Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66, 1100–1103, https://doi.org/10.1099/ijsem.0.000760 (2016).
Acknowledgements
This work was supported by the Korea Institute of Marine Science and Technology Promotion (KIMST) (KIMST Grant 20220632; KOPRI-PM24050).
Author information
Authors and Affiliations
Contributions
J.J. and Y.M.L. conceptualized and designed the study. Y.P., D.H.L., J.H.K., Y.K.J., J.K.H. and Y.M.L. collected the samples. J.J., Y.P. and Y.M.L. performed bioinformatics analyses, prepared figures and tables, and wrote the original draft. Y.K.J. and J.K.H. provided the funding. All authors reviewed, edited, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jeon, J., Park, Y., Lee, DH. et al. Microbial profiling of the East Siberian Sea sediments using 16S rRNA gene and metagenome sequencing. Sci Data 11, 1350 (2024). https://doi.org/10.1038/s41597-024-04177-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-04177-0
