
Abstract
The growing popularity of machine learning (ML) and deep learning (DL) in scientific fields is hindered by the scarcity of high-quality datasets. While quantum mechanical (QM) predictions using DL techniques such as graph neural networks (GNNs) and generative models are gaining traction, insufficient training data remains a bottleneck. The QM40 dataset addresses this challenge by representing 88% of the FDA-approved drug chemical space. It includes molecules containing 10 to 40 atoms and composed of elements commonly found in drug molecular structures (C, O, N, S, F, Cl). QM40 offers valuable resources for researchers which include the core QM40 main dataset, containing 16 key quantum mechanical parameters for 162,954 molecules calculated using the B3LYP/6-31G(2df,p) level of theory in Gaussian16, ensuring consistency with established datasets like QM9 and Alchemy. This compatibility allows for future concatenation of QM40 with these datasets. In addition to other valuable information, the QM40 dataset offers the initial and optimized Cartesian coordinates, Mulliken charges, and detailed bond information, including local vibrational mode force constants, which serve as indicators of bond strength. QM40 can be used to benchmark both existing and new methods for predicting QM calculations using ML and DL techniques.
Similar content being viewed by others
Background & Summary
History demonstrates that access to high-quality, well-organized data significantly advances specific fields. The ImageNet1 dataset exemplifies this perfectly. It provided a benchmark dataset for image classification and supported introducing groundbreaking architectures such as AlexNet2, VGG3, and ResNet4. Electronic structure and property calculations have become essential in modern materials and drug discovery research and development (R&D) portfolios. While quantum mechanical (QM) methods like Coupled Cluster (CC), Multi-configurational self-consistent field (MCSCF), etc offer the highest accurate data but are computationally intensive. Density Functional Theory (DFT) offers a better compromise between accuracy and efficiency. However, its computational requirements still make it unsuitable for large-scale drug screening. A central challenge in modern theoretical chemistry is to develop and implement approximations that accelerate QM methods while maintaining accuracy. Recent advances in machine learning (ML) techniques have proven immensely useful to address this challenge. ML can either minimize the need for extensive QM calculations or even bypass them altogether5. However, the performance of ML methods, including graph neural networks (GNNs)6,7, large language models (LLMs)8,9, and generative models10, is heavily influenced by the size and quality of the training data. The ability of currently available QM datasets to provide the size and quality required for machine learning applications is questionable. We believe developing a high-quality dataset will catalyze the application of ML techniques for predicting QM properties, coordinates and bond strengths.
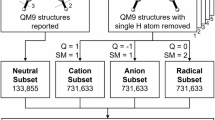
Despite its widespread use in drug discovery and materials science, the so-called QM911 dataset has limitations. Composed solely of smaller molecules with a maximum of nine atoms (C, O, N, and F), it fails to represent the full spectrum of chemical complexity in real-world applications, particularly drug discovery, where molecules are often much larger. While QMugs12 offers the advantage of a vast collection of drug-like molecules (over 665,000) and the ability to handle structures with up to 100 atoms, it’s important to consider that these molecules were optimized using a less computationally expensive, but potentially less accurate, semi-empirical level of theory. Table 1 describes the selected QM datasets currently available. Additionally, analyzing 2600 FDA-approved drugs13 we found that the QM9 molecules capture only 10% of drug-relevant space, while the molecules with 40 atoms encompass 88 % as illustrated in Fig. 1b. Therefore, we have designed the QM40 database, which considerably expands the QM9 chemical space by incorporating molecules with up to 40 atoms including also S and Cl (C, O, N, S, F, and Cl), making it a valuable training set for ML tasks predicting various QM parameters as depicted in Fig. 1a. The QM40 dataset includes 162,954 molecules originally obtained from the ZINC14 dataset which contains nearly 700 million drug-like molecules.

Statistical analysis of 2,584 FDA-approved drugs by (DrugCentral 2023)44 (a) Distribution of heavy atoms, (b) Distribution of heavy atom count.
QM calculations are performed at the B3LYP/6-31G(2df,p) level of theory in consistency with the QM9 and Alchemy datasets. The computational method was chosen to provide the best compromise between accuracy and efficiency, following recent suggestions in the literature15,16,17. Additionally, QM40 can be seamlessly combined with QM9, which includes molecules with 0-10 heavy atoms, while QM40 covers molecules with more than 10 heavy atoms, as both datasets were generated using the same method. In particular, QM40 offers a new feature, including our unique local vibrational mode force constant as a quantitative bond strength measure18,19. Normal vibrational modes are generally delocalized due to kinematic and electronic coupling20,21. A certain normal vibrational mode cannot always be associated with an isolated bond because it can combine with other molecular fragment stretching, bending, or torsional movements. This combination hinders the direct relationship between the normal stretching frequency or associated normal mode force constant and bond strength and the comparison between stretching modes in related molecules. Konkoli and Cremer addressed this problem by solving mass-decoupled Euler–Lagrange equations22,23,24 and introducing the Local Vibrational Mode Theory. In particular, the local mode force constants ka have qualified as a quantitative measure of bond strength for both covalent bonds25,26,27,28 and weak chemical interactions29,30,31. The QM40 dataset is continuously updated with additional molecules and features. New information can be found on our Figshare repository32 and GitHub page QM40 dataset for ML.
In the dataset descriptor list reported here, we provide an extended dataset beyond QM9, accommodating up to 40 heavy atoms, which represents 88% of the FDA-approved drug chemical space, thus offering a closer reflection of drug-like chemical space. Additionally, It includes bond strength data for all bonds within the dataset. Therefore, we anticipate that the QM40 dataset will establish itself as a new standard benchmark for evaluating current and future methods in machine-learned potentials. Even more significantly, it is a robust foundation for developing future general-purpose machine-learned potentials. This dataset provides a substantial head start on data generation, and its capabilities can be further enhanced by incorporating existing or future datasets encompassing additional relevant regions of chemical space.
Methods
QM calculations
All electronic structure calculations, including geometry optimizations and frequency calculations, were carried out using the B3LYP/6-31G(2df,p) level of theory in the Gaussian1633 package. Local mode force constants were calculated with our LModeA34 software package and local vibrational mode parameters were automatically generated using our LModeAGen protocol35.
Molecular geometry generation
The QM40 dataset is a meticulously chosen subset of molecules from the ZINC database, specifically designed for drug discovery applications. To achieve this focus, QM40 excludes anions and cations and only considers neutral molecules with a maximum of 40 atoms composed of C, N, O, S, F, and Cl. This selection of atom count and elements aligns with the analysis of FDA-approved drugs up to 2023. Figure 1 depicts the distribution of atom count (a), and elements (b) in FDA-approved drugs.
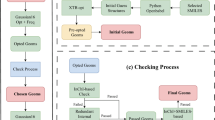
Molecular SMILES strings from the ZINC database were converted into PDB files using RDKit36. This process incorporates atomic connectivity, atomic coordinates, and the addition of hydrogen atoms, resulting in charge-neutral singlet ground states. The initial geometries for DFT calculations were obtained by pre-optimizing the structures using the extended tight-binding (xTB)37 method with the GFN2-xTB38 level of theory. Employing the final optimized coordinates from the xTB calculations, DFT calculations were performed, followed by frequency calculations. LModeA calculations were performed for each molecule using the final checkpoint file generated from the corresponding frequency calculation. Any molecule encountering convergence failures, imaginary frequencies, or LModeA unphysical parameters was excluded from the dataset throughout each stage. Figure 2 comprehensively illustrates the data generation workflow.

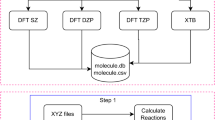
Scheme for generating optimized QM parameters, geometry and Local vibrational mode frequencies of 162,954 molecules from the ZINC database.
Data Records
The QM40 dataset is archived in CSV file format and publicly available through a Figshare data repository32. The dataset is organized into three separate sets of CSV files. The core information resides in the “QM40 Main Dataset” CSV file containing 162,954 SMILES strings and corresponding QM parameters. These parameters are detailed in Table 2. “QM40 xyz Dataset” stores each molecule’s initial and optimized atomic Cartesian coordinates alongside Mulliken charges. The third file, “QM40 bond Dataset” contains the bond information with local mode force constants for every bond within the molecule. The QM40 xyz and bond datasets are further detailed in Tables 3 and 4, respectively.
Technical Validation
Validation of geometric consistency
The geometry optimization of structures initially derived from SMILES strings can lead to changes that alter the type of molecule, causing inconsistencies between the optimized geometry and the original SMILES code. To address this, the consistency of the B3LYP optimization in the dataset was verified using LModeA to check for unphysical parameters. LModeA input files were generated based on connectivity information derived from the initial geometry in the PDB files. The LModeA package then uses this connectivity information to retrieve optimized data from the formatted checkpoint file (FCHK) for local vibrational mode analysis. If the specific connectivity in the LModeA input file does not match that created from the optimized FCHK file, the LModeA package returns the message, “Unphysical parameter was detected. Molecules with unphysical parameters were selectively removed from the dataset, as they represent conformers that do not correspond to the original molecule. Figure 2 provides a graphical representation of this procedure.
Validation of quantum chemistry results
We modeled all 162,954 molecules using the B3LYP/6-31G(2df,p) level of DFT. This approach aligns with the methodology used for the QM9 dataset. We specifically focused on molecules containing more than 10 atoms. This allows for the concatenation of QM9 with QM40, creating a combined dataset with approximately 300k molecules. The chosen B3LYP/6-31G(2df,p) level has been previously validated against high-level theories (G4MP2, G4, and CBS-QB3) used in the QM9 study.
Validation of the QM40 chemical space
The chemical space of QM40 was validated using two methods. The first method involved dividing the QM40 dataset into six classes based on the number of atoms per molecule: 10-15, 15-20, 20-25, 25-30, 30-35 and 35-40. The number of molecules in each class was then calculated and visualized in Fig. 3. As shown in the figure, nearly 26% of the molecules belong to the 10-15 atom range, followed by 21% in the 25-30 atom range. The 20-25 atom range has the smallest representation, at 7%. It’s important to note that despite these variations, all classes contain over 12,000 molecules.

Number of heavy atom composition of QM40 dataset.
To further validate the chemical space of QM40, the dataset was split into 16 distinct databases based on specific bond types (CC, CH, OH, NH, etc., detailed in Table 5). For each bond type, we calculated the number of molecules containing that bond, the total number of such bonds, and the maximum, minimum, average, and standard deviation of the local vibrational stretching force constant. This analysis confirms the consistency of the data concerning the presence of different bond types in the geometries of the dataset. It also verifies that all bonds were formed exclusively by the elements C, O, N, S, Cl, F, and H. Furthermore, the top three maximum bond strengths were identified in NN triple bonds, CN triple bonds, and CC triple bonds, consistent with their experimental bond dissociation enthalpy values27,39. Conversely, the analysis revealed a low prevalence of SS, NF, and SH bonds in the QM40 dataset, suggesting a natural scarcity of these bond types in drug-like compounds40.
Usage Notes
QM40 provides a GitHub repository. The repository includes a user-friendly Python application for generating the dataset. This application can be easily installed using common pip package managers. In addition, the repository offers a Python module specifically designed for interacting with the QM40 data users. This module provides functionalities for navigating the QM properties, geometries and bond information, extracting specific information, and even downloading subsets of interest. To ensure smooth exploration and utilization, it comes with a README file and tutorials that detail technical specifications and include usage examples.
Code availability
QM40 GitHub repository can be accessed and downloaded under CC BY 4.0 license (QM40 dataset for ML). Additionally, the QM40 website is available online (QM40 website).
References
Deng, J. et al. Imagenet: A large-scale hierarchical image database. In 2009 IEEE conference on computer vision and pattern recognition, 248–255 (Ieee, 2009).
Krizhevsky, A., Sutskever, I. & Hinton, G. E. Imagenet classification with deep convolutional neuralnetworks. Adv.neural information processing systems 25 (2012).
Simonyan, K. & Zisserman, A. Very deep convolutional networks for large-scale imagerecognition. Int. Conf. on Learn. Represent. (2015).
He, K., Zhang, X., Ren, S. & Sun, J. Deep residual learning for image recognition. In Proceedings of the IEEE conference on computer vision and pattern recognition, 770–778 (2016).
Chandrasekaran, A. et al. Solving the electronic structure problem with machine learning. Npj Comput. Mater. 5, 22 (2019).
Xiong, J., Xiong, Z., Chen, K., Jiang, H. & Zheng, M. Graph neural networks for automated de novo drug design. Drug Discov. Today 26, 1382–1393 (2021).
Zhang, Z. et al. Graph neural network approaches for drug-target interactions. Curr. Opin. Struct. Biol. 73, 102327 (2022).
Chakraborty, C., Bhattacharya, M. & Lee, S.-S. Artificial intelligence enabled chatgpt and large language models in drug target discovery, drug discovery, and development. Mol. Ther. Nucleic Acids 33, 866–868 (2023).
Pal, S., Bhattacharya, M., Islam, M. A. & Chakraborty, C. Chatgpt or llm in next-generation drug discovery and development: pharmaceutical and biotechnology companies can make use of the artificial intelligence-based device for a faster way of drug discovery and development. Int. J. Surg. 109, 4382–4384 (2023).
Tong, X. et al. Generative models for de novo drug design. J. Med. Chem. 64, 14011–14027 (2021).
Ramakrishnan, R., Dral, P. O., Rupp, M. & Von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. data 1, 1–7 (2014).
Isert, C., Atz, K., Jiménez-Luna, J. & Schneider, G. Qmugs, quantum mechanical properties of drug-like molecules. Sci. data 9, 273 (2022).
of Medicine at University of New Mexico, S. Drug central 2023. https://drugcentral.org/ (Last updated: (Sep 09 2023)). Accessed: (Apr 15 2024).
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S. & Coleman, R. G. Zinc: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768 (2012).
Berezin, K. & Nechaev, V. Comparison of theoretical methods and basis sets for ab initio and dft calculations of the structure and frequencies of normal vibrations of polyatomic molecules. J. Appl. Spectrosc. 71, 164–172 (2004).
Curtiss, L. A., Redfern, P. C. & Raghavachari, K. Gaussian-4 theory using reduced order perturbation theory. J. Chem. Phys.127 (2007).
Burk, P., Koppel, I. A., Koppel, I., Leito, I. & Travnikova, O. Critical test of performance of b3lyp functional for prediction of gas-phase acidities and basicities. Chem. Phys. Lett. 323, 482–489 (2000).
Kraka, E., Quintano, M., La Force, H. W., Antonio, J. J. & Freindorf, M. The Local Vibrational Mode Theory and Its Place in the Vibrational Spectroscopy Arena. J. Phys. Chem. A 126, 8781–8900 (2022).
Kraka, E., Zou, W. & Tao, Y. Decoding chemical information from vibrational spectroscopy data: Local vibrational mode theory. WIREs: Comput. Mol. Sci. 10, 1480 (2020).
Wilson, E. B., Decius, J. C. & Cross, P. C. M.Molecular Vibrations. The Theory of Infrared and Raman Vibrational Spectra (McGraw-Hill, New York, 1955).
Kelley, J. D. & Leventhal, J. J. In Problems in Classical and Quantum Mechanics: Normal Modes and Coordinates, 95–117 (Springer, 2017).
Konkoli, Z., Larsson, J. A. & Cremer, D. A New Way of Analyzing Vibrational Spectra. II. Comparison of Internal Mode Frequencies. Int. J. Quantum Chem. 67, 11–27 (1998).
Konkoli, Z. & Cremer, D. A New Way of Analyzing Vibrational Spectra. III. Characterization of Normal Vibrational Modes in terms of Internal Vibrational Modes. Int. J. Quantum Chem. 67, 29–40 (1998).
Konkoli, Z., Larsson, J. A. & Cremer, D. A New Way of Analyzing Vibrational Spectra. IV. Application and Testing of Adiabatic Modes within the Concept of the Characterization of Normal Modes. Int. J. Quantum Chem. 67, 41–55 (1998).
Delgado, A. A. A., Humason, A., Kalescky, R., Freindorf, M. & Kraka, E. Exceptionally Long Covalent CC Bonds - A Local Vibrational Mode Study. Molecules 26, 950–1–950–25 (2021).
Kraka, E., Larsson, J. A. & Cremer, D. Generalization of the Badger Rule Based on the Use of Adiabatic Vibrational Modes. In Grunenberg, J. (ed.) Computational Spectroscopy, 105–149 (Wiley, New York, 2010).
Kalescky, R., Kraka, E. & Cremer, D. Identification of the Strongest Bonds in Chemistry. J. Phys. Chem. A 117, 8981–8995 (2013).
Kraka, E. & Cremer, D. Characterization of CF Bonds with Multiple-Bond Character: Bond Lengths, Stretching Force Constants, and Bond Dissociation Energies. ChemPhysChem 10, 686–698 (2009).
Freindorf, M., Yannacone, S., Oliveira, V., Verma, N. & Kraka, E. Halogen Bonding Involving I2 and d8 Transition-Metal Pincer Complexes. Crystals 11, 373–1–373–21 (2021).
Kalescky, R., Zou, W., Kraka, E. & Cremer, D. Local Vibrational Modes of the Water Dimer - Comparison of Theory and Experiment. Chem. Phys. Lett. 554, 243–247 (2012).
Kalescky, R., Kraka, E. & Cremer, D. Local Vibrational Modes of the Formic Acid Dimer - The Strength of the Double H-Bond. Mol. Phys. 111, 1497–1510 (2013).
Kalapuwage, A. M. M. Qm40: A more realistic qm dataset for machine learning in molecular science. Figshare https://doi.org/10.6084/m9.figshare.25993060.v1 (2024).
Frisch, M. J. et al. Gaussian ~ 16 Revision C.01 (2016).
Zou, W. et al. LModeA2023. Computational and Theoretical Chemistry Group (CATCO), Southern Methodist University: Dallas, TX, USA (2023).
Moura Jr, R. T., Quintano, M., Antonio, J. J., Freindorf, M. & Kraka, E. Automatic Generation of Local Vibrational Mode Parameters: From Small to Large Molecules and QM/MM Systems. J. Phys. Chem. A 126, 9313–9331 (2022).
RDKit. Rdkit: Open-source cheminformatics http://www.rdkit.org (2023).
Bannwarth, C. et al. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 11, e1493 (2021).
Bannwarth, C., Ehlert, S. & Grimme, S. Gfn2-xtb—an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 15, 1652–1671 (2019).
Luo, Y.-R. Comprehensive handbook of chemical bond energies (CRC press, 2007).
Xu, J. & Stevenson, J. Drug-like index: a new approach to measure drug-like compounds and their diversity. Journal of Chemical Information and Computer Sciences 40, 1177–1187 (2000).
Ramakrishnan, R., Hartmann, M., Tapavicza, E. & Von Lilienfeld, O. A. Electronic spectra from tddft and machine learning in chemical space. J. Chem. Phys.143 (2015).
Chen, G. et al. Alchemy: A quantum chemistry dataset for benchmarking ai models. Int. Conf. on Learn. Represent. (2019).
Smith, J. S., Isayev, O. & Roitberg, A. E. Ani-1, a data set of 20 million calculated off-equilibrium conformations for organic molecules. Sci. data 4, 1–8 (2017).
Avram, S. et al. Drugcentral 2023 extends human clinical data and integrates veterinary drugs. Nucleic Acids Res. 51, D1276–D1287 (2023).
Acknowledgements
This work was financially supported by the National Science Foundation under grant number CHE 2102461. We also acknowledge the computational resources provided by the O’Donnell Data Science and Research Computing Institute at Southern Methodist University. MRJ also thanks the Brazilian National Council for Scientific and Technological Development - CNPq, Grant numbers 406483/2023-0, and 310988/2023-3.
Author information
Authors and Affiliations
Contributions
All authors conceived of the presented idea. A.M. implemented the methods and carried out calculations. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Madushanka, A., Moura, R.T. & Kraka, E. QM40, Realistic Quantum Mechanical Dataset for Machine Learning in Molecular Science. Sci Data 11, 1376 (2024). https://doi.org/10.1038/s41597-024-04206-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04206-y
