
Abstract
Compared with leaner breeds, local Chinese pig breeds have distinct intestinal microbial, as determined by metagenomic techniques, and the interactions between oral microorganisms and their hosts are also gradually being clarified. However, the high host genome content means that few metagenome-based oral microbiomes have been reported. Here, we combined dilution-based metagenomic sequencing and binning approaches to extract the microbial genomes from the oral microbiomes of Tibetan and Duroc pigs. The host contamination rates were reduced to 13.64%, a quarter of the normal metagenomic level (65.25% on average). Medium–high-quality metagenome-assembled genomes (MAGs; n = 3,448) spanning nine phyla were retrieved and 70.79% were novel species. Of the nonredundant MAGs, only 13.37% were shared, revealing the strong disparities between Tibetan and Duroc pigs. The oral microbial diversity of the Duroc pig was greater than that of the Tibetan pig. We present the first large-scale dilute-based metagenomic data on the pig oral microbiome, which should facilitate further investigation of the functions of oral microorganisms in pigs.
Similar content being viewed by others

Background & Summary
The oral microbiota is considered the second largest microbiota after the intestinal microbiota1,2,3. Because it is a complex environment, the animal oral cavity provides suitable conditions for the colonization and proliferation of various microorganisms, including an ideal temperature, humidity, and nutrient sources. Under normal circumstances, the microorganisms in the oral cavity maintain a balance with the body, in terms of both the quantities and types of microbes. An imbalance in the oral microbiota can lead to various oral diseases, such as dental caries, periodontitis, gingivitis, oral ulcers, and oral cancer4,5,6. The interactions between and changes in oral microbial communities correlate significantly with oral diseases and systemic diseases, which significantly affect animal health7,8.
The Tibetan pig (TP) is a local breed in China that has lived for a long time in unpolluted, natural mountainous areas, adapting to harsh climatic conditions at high altitudes9,10. Compared with leaner pig breeds, it shows strong adaptability, stress resistance, disease resistance, and tolerance of coarse feed11. It has tender meat, a delicious taste, unique flavor, and less fat and more lean meat than other breeds, and therefore better meets the current requirement for high-quality pork9. However, it has the disadvantages of a slow growth rate and small litter size. The Duroc pig (DP) is a lean meat pig breed, with a large breeding population all over the world, and has the advantages of rapid growth, a high feed conversion rate, and high percentage of lean meat12. Recent studies have shown that there are significant differences in the intestinal microbiotas of local Chinese pig breeds and commercialized breeds13. Metagenomic binning approaches were applied in oral microbiome research and superior approach such as multi-coverage binning strategy was also applied to retrieve more comprehensive binning results14,15. In addition to updating data analysis pipelines, the microfluidics-based mini-metagenomics strategy could also be a powerful tool for dissecting microbial community structure in complex habitats16. However, few metagenomic analyses of the pig oral microbiome have been reported because oral metagenome data are strongly contaminated with the host genome.
Here, we first used the dilution-based metagenomic sequencing (Dilute-Meta-Seq) approach (Fig. 1), which combines the dilute-based metagenomic sequencing technology and metagenome-assembled genome (MAG) binning approach to investigate and compare the oral microbiome community structures of TP and DP. Briefly, the first step was diluting the liquid suspension with microbial cells and preparing enough DNA from different dilution gradients. Then whole-genome metagenomic sequencing was introduced to do MAG binning. Our results demonstrate the great power of the Dilute-Meta-Seq method for samples with high host contamination in metagenomic studies. In addition we also assembled 200 Dilute-Meta-Seq samples and two whole-genome shotgun (WGS) samples, and retrieved 3,448 medium–high-quality MAGs spanning nine bacterial phyla in total. To our knowledge, this is the first study to undertake large-scale metagenomic sequencing of the pig oral microbiome, extracting thousands of oral MAGs. This rich oral MAG resource should provide a basic scientific reference with which to study the physiological effects of the oral microbiota in different pig breeds.

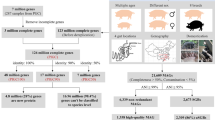
Workflow of Dilute-Meta-Seq and whole-genome shotgun (WGS) sequencing of the oral microbiome of pigs (https://www.biorender.com/, agreement number: QM26O4UX6B). Multiple displacement amplification (MDA) is one of whole-genome amplification methods for small amounts of DNA samples.
Methods
Sample collection
In this study, we collected oral microbiotal samples from six adult TP boars and six adult DP boars. All animal procedures were approved by Yunnan Agricultural University’s Life Science Ethics Committee (202309030) and were carried out in accordance with the Guidelines on the Humane Treatment of Laboratory Animals (approval no. 2006-398). To minimize the diurnal variations in saliva flow and composition, all saliva samples were collected at 12 noon. Before the comprehensive oral examination of all the subjects, the pigs were fasted for at least 1 h. In contrast to the collection of human saliva, pigs were not subject to a gargling procedure. Instead, unstimulated whole saliva samples were collected from each subject with a sterile cotton swab17 and stored in a 5 ml sterile DNA-free conical tube. All collected samples were immediately snap-frozen in liquid nitrogen and stored at −80 °C.
DNA preparation for dilute-meta-seq
The saliva samples were eluted with 1 ml of precooled 1 x phosphate-buffered saline (PBS; maintained at 4 °C). The six liquid samples from pigs of the same breed were combined. The 6 ml volume of the precooled 1 x PBS solution was transferred to a 10 ml centrifuge tube. The samples were filtered through a 40 µm cell strainer (catalogue: 27305; STEMCELL Technologies, Shanghai China) and centrifuged at 5000 × g for 10 min to remove the host tissue residue. The filtered solution was used in subsequent experimental procedures.
To determine the dilution factor for the filtered solution of enriched microbial cells, a four-member dilution gradient from 1e−2 to 1e−5 was established in PBS. The cell counter QUANTOM Tx™ (Logos Biosystems, South Korea) was used to confirm the suitable dilution gradient. In this study, we selected a 1e−3 × PBS dilution strategy because there were about 50–200 microbial cells in each tube at this dilution. To maximize the number of binned MAGs, a total of 200 diluted samples (100 for TP and 100 for DP) at the appropriate dilution were extracted from the raw filtered solutions. For each diluted sample, the REPLI-g Single Cell Kit (catalogue: 150343; Qiagen) was used to produce enough genomic DNA for further library construction and sequencing with whole-genome amplification (WGA). This kit uses gentle alkaline incubation to lyse microbial cells and release low concentrations of DNA fragments and an optimized Phi 29 polymerase formulation to complete the multiple-displacement amplification procedure. At least 1 µg of WGA DNA was finally produced for each diluted sample.
To evaluate the contamination ratio for the pig host genome in each diluted sample, a quantitative PCR analysis of the pig beta-actin gene (ACTB) was performed with QK Platinum SYBR Green Master Mix (catalogue: A57156; Thermo Fisher Scientific). The forward primer sequence was 5′-GGCATCGTGATGGACTCCG-3′ and the reverse primer sequence was 5′-GCTGGAAGGTGGACAGTGAG-3′. Each 20 μl reaction contained 2 μl of MGA DNA and 5 pmol of each primer. Real-time qPCR was run on the SLAN-96P Real-Time PCR System (Hongshitech, China). The cycling conditions were one cycle of denaturation at 95 °C for 10 min, followed by 40 three-segment cycles of amplification (95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s), during which fluorescence was automatically measured, and one three-segment cycle of product melting (95 °C for 1 min, 55 °C for 30 s, and 95 °C for 30 s). A diluted sample with a cycle threshold (Ct) value of >25 was considered a successful Dilute-Meta-Seq sample because the host genome ratio was low. Failed samples were discarded and the above process repeated until sufficient samples (100 for each breed) were generated.
DNA extraction for WGS and sequencing
Microbial DNA for WGS sequencing was extracted from enriched microbial cell suspensions isolated from the TP and DP saliva samples. The Blood & Tissue DNA Extraction Kit (catalogue: M6399-00; Omega Biotek, USA) was used for cell lysis and DNA extraction. For the two WGS samples and 200 WGA samples, 500 ng of DNA per sample was required for metagenomic library construction. The metagenomic DNA library for each sample was constructed with the NEBNext® Ultra™ II DNA Library Prep Kit (catalogue: E7645L; New England Biolabs), according to the manufacturer’s instructions. The length of the inserted fragments (approximately 300 bp) and the DNA concentrations of all libraries were evaluated with the Agilent 2100 Bioanalyzer. Then the sequencing libraries were subjected to high-throughput sequencing on the MGI DNBSEQ-T7 system in 150 bp pair-end mode.
Metagenomic assembly and binning
The sequencing raw reads were quality trimmed with Trimmomatic v0.3318, with the options ‘SLIDINGWINDOW:4:15 MINLEN:75’, and were mapped against the pig genome sequence (National Center for Biotechnology Information accession: GCF_000003025.6) using the ‘mem’ module in BWA v0.7.17-r118819 to calculate the host genome contamination ratio. Clean reads were considered as those trimmed reads with no pig genome, and were assembled separately for each sample with MEGAHIT v1.1.120. Only contigs with a length of >500 bp were retained. The sequencing depth of each contig was determined by mapping reads from the corresponding sample using the ‘jgi_summarize_bam_contig_depths’ script in the MetaBAT2 v2.11.121 package. The binning analysis was performed with MetaBAT2 Genome completeness and contamination were estimated with CheckM2 v1.0.222. The quality of the MAGs was determined based on the MIMAG23 standard and only those of high- (≥90% completeness, ≤5% contamination) or medium quality (≥50% and <90% completeness, >5% and <10% contamination) were retained for downstream analysis. Ribosomal RNA genes (rRNA) were predicted with barrnap v0.9 (https://github.com/tseemann/barrnap)24 with domain-specific models. Transfer RNA genes were predicted with aragorn v1.2.3825 with the default parameters.
MAG taxonomic classification and dereplication
The taxonomy of the MAGs was determined with the Genome Taxonomy Database toolkit (GTDB-Tk)26, database version R214, and toolkit version v1.9.0. To infer the phylogenetic positions of the studied MAGs, phylogenomic trees were constructed based on bacterial sequence alignments of 400 universal marker genes, constructed with PhyloPhlAn v3.027. The phylogenomic trees were visualized with iTOL28. Redundant MAGs were identified with dRep v3.2.029 with the option ‘-pa 0.95’, which sets the average nucleotide identity (ANI) to 95%, and the MAGs with the highest quality scores (QS = completeness – [5 × contamination]) were taken as the representative MAGs. Finally, the clean reads were mapped to the total representative MAGs with the ‘salmon’ tool30 in the metaWRAP31 package. The relative abundances and read recruitment ratios of the MAGs were calculated from the ‘salmon’ output.
Technical Validation
Two hundred Dilute-Meta-Seq samples were sequenced (100 for DP and 100 for TP), and 5.38 billion raw reads (26.92 million raw reads/sample on average) and 4.46 billion clean reads (22.29 million clean reads/sample on average) were produced in total (Table S1, Fig. 2A). The WGS method was included in this study to evaluate the performance of the Dilute-Meta-Seq workflow (Fig. 1). We generated 224.25 and 329.64 million reads from the DP and TP oral microbiome samples, respectively. And the nucleic acids with sequencing accuracy above 99.9% (also called Q30 values) was 94.70% on average (Table S1). The average host genome ratio for the Dilute-Meta-Seq samples was 13.64% (DP: 23.67% ± 5.65%; TP: 3.61% ± 4.79%), whereas the host ratio for the TP Dilute-Meta-Seq samples was much lower than that of the DP samples. The host ratios of the WGS samples were 57.43% and 73.06% for DP and TP, respectively (Fig. 2B). The 200 Dilute-Meta-Seq metagenomes were assembled separately into 25.55 million contigs, totaling 36.70 Gb (ranging from 5.52 to 468.86 Mb for each sample) and the average metagenomic assembly sizes were 195.60 and 171.41 Mb for DP and TP, respectively, according to MEGAHIT (Table S1, Fig. 2C,D). The metagenomic assembly sizes were 701.94 and 238.13 Mb for the DP and TP WGS samples, respectively.

Read content and assembly status of Dilute-Meta-Seq data. (A) Distribution of raw and clean data (with low-quality reads and host genome reads removed) for the Duroc pig (DP) and Tibetan pig (TP) Dilute-Meta-Seq samples. Host contamination ratio (B), number of contigs (C) and contig size (D) for the DP and TP Dilute-Meta-Seq samples. Asterisks indicate the status of the corresponding whole-genome shotgun (WGS) samples.
We retrieved 8,815 MAGs from the 200 Dilute-Meta-Seq samples (8,601) and two WGS samples (214) with MetaBAT2. Of these MAGs, 7.69% (678/8,815) were considered high-quality MAGs (≥90% completeness and ≤5% contamination) and 31.42% (2,770/8,815) were considered medium-quality MAGs (≥50% completeness and <10% contamination), according to CheckM v2 (Table S2). The genome sizes of these 3,448 medium–high-quality MAGs ranged from 0.34 to 8.75 Mbp, with GC contents varying from 29% to 73%. Furthermore, 533 (15.46%) MAGs were predicted to contain at least one copy of the 16S rRNA gene (Table S2). The 3,448 medium–high-quality MAGs were clustered into 202 representative species-level groups by dRep, based on 95% ANI, and 702 representative strain-level MAGs were classified with ANI >99% (Table S2).
A phylogenetic analysis of 400 microbial universal marker genes showed that the 202 representative species-level MAGs belonged to nine bacterial phyla, according to the GTDB taxonomy (Fig. 3). In the pig oral microbiome ecosystem, bacterial species were dominated by members (n > 5) of the Bacteroidota (n = 65), Pseudomonadota (n = 51), Bacillota_A (n = 31), Bacillota (n = 24), Patescibacteria (n = 11), Actinomycetota (n = 8) and Fusobacteriota (n = 7) (Fig. 3, Table S2). Of these 202 MAGs, 20 represented novel genera and 142 novel species. The relative taxonomic abundances differed between the DP and TP WGS samples (correlation coefficient, R2 = 0.14; Table S3, Fig. 3).

Phylogeny of 202 representative species-level metagenome-assembled genomes (MAGs) based on a set of 400 conserved prokaryotic marker genes. Phyla are color-coded and taxonomy is from the Genome Taxonomy Database (GTDB). Concentric rings moving outward from the tree show the completeness, genome size, and number of assembled MAGs of these 202 MAGs. The outermost layer indicates the relative abundances of MAGs within the Duroc pig (DP) and Tibetan pig (TP) whole-genome shotgun (WGS) samples.
We then characterized the 202 representative species-level MAGs identified in this study (Fig. 4A). Of the nonredundant MAGs, 13.37% (27/202) were shared by both the DP and TP samples. Although the number of nonredundant MAGs retrieved with the Dilute-Meta-Seq approach became saturated at sampling numbers exceeding 75 (Fig. 4B), the WGS samples still generated 22.8% unique species. The average read recruitment ratios for the 202 representative species-level MAGs were 70.40% and 67.40% for the DP and TP Dilute-Meta-Seq samples, respectively. These results show that the majority of reads aligned to the 202 representative species-level MAGs and the high completeness of our MAGs binning data in pig oral ecosystem.

Metagenome-assembled genome (MAG) characteristics in the different samples and experimental strategies. (A) Number of representative species-level MAGs shared among Dilute-Meta-Seq and whole-genome shotgun (WGS) samples. (B) Plots of the saturation curves illustrate the increases in the numbers of MAGs together with the Dilute-Meta-Seq sample growth. (C) Distribution of the read recruitment ratios for Dilute-Meta-Seq samples.
Code availability
Custom scripts were not used to process this dataset. And the software versions and non-default parameters used in this study had been described in the methods sections.
References
Zhang, Y. et al. Human oral microbiota and its modulation for oral health. Biomed Pharmacother. 99, 883–893 (2018).
Verma, D., Garg, P. K. & Dubey, A. K. Insights into the human oral microbiome. Archives of microbiology. 200, 525–540 (2018).
Gao, L. et al. Oral microbiomes: more and more importance in oral cavity and whole body. Protein & cell. 9, 488–500 (2018).
Maier, T. Oral Microbiome in health and disease: maintaining a healthy, balanced ecosystem and reversing dysbiosis. Microorganisms. 11, 1453 (2023).
Wade, W. G. The oral microbiome in health and disease. Pharmacological research. 69, 137–143 (2013).
Gomez, A. et al. Host genetic control of the oral microbiome in health and disease. Cell host & microbe. 22, 269–278 (2017).
Jorth, P. et al. Metatranscriptomics of the human oral microbiome during health and disease. MBio. 5, 10–1128 (2014).
Sampaio-Maia, B., Caldas, I. M., Pereira, M. L., Pérez-Mongiovi, D. & Araujo, R. The oral microbiome in health and its implication in oral and systemic diseases. Advances in applied microbiology. 97, 171–210 (2016).
Gan, M. et al. High altitude adaptability and meat quality in Tibetan pigs: A reference for local pork processing and genetic improvement. Animals. 9, 1080 (2019).
Ai, H. et al. Population history and genomic signatures for high-altitude adaptation in Tibetan pigs. BMC genomics. 15, 1–14 (2014).
Shang, P., Wei, M., Duan, M., Yan, F. & Chamba, Y. Healthy gut microbiome composition enhances disease resistance and fat deposition in Tibetan pigs. Frontiers in Microbiology. 13, 965292 (2022).
Kim, J. A. et al. The effects of breed and gender on meat quality of Duroc, Pietrain, and their crossbred. J Anim Sci Technol. 62, 409 (2020).
Zhao, F. et al. Gut microbiome signatures of extreme environment adaption in Tibetan pig. NPJ Biofilms Microbiomes. 9, 27 (2023).
Mattock, J. & Watson, M. A comparison of single-coverage and multi-coverage metagenomic binning reveals extensive hidden contamination. Nature Methods. 20, 1170–1173 (2023).
Valles-Colomer, M. et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature. 614, 125–135 (2023).
Yu, F. B. et al. Microfluidic-based mini-metagenomics enables discovery of novel microbial lineages from complex environmental samples. Elife. 6, e26580 (2017).
Murase K. et al. Characterization of pig saliva as the major natural habitat of Streptococcus suis by analyzing oral, fecal, vaginal, and environmental microbiota. PLoS One. e0215983 (2019).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–20 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–60 (2009).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 31, 1674–6 (2015).
Kang, D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. Peer J. 26, e7359 (2019).
Chklovski, A., Parks, D. H., Woodcroft, B. J. & Tyson, G. W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods. 20(8), 1203–1212 (2023).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nature biotechnology. 36, 660 (2017).
Karin, L. et al. Rnammer: consistent and rapid annotation of ribosomal rna genes. Nucleic Acids Research. 35, 3100 (2007).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–6 (2004).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 36, 1925–1927 (2019).
Asnicar, F. et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun. 11, 2500 (2020).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic acids research. 49, W293–W296 (2021).
Olm, M. R. et al. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. The ISME Journal. 11, 2864–2868 (2017).
Patro, R. et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. Apr;14(4):417–419 (2017).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 6, 158 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP501462 (2024).
Figshare https://doi.org/10.6084/m9.figshare.24947385.v1 (2024).
NCBI GenBank https://identifiers.org/ncbi/bioproject:PRJNA1098453 (2024).
Acknowledgements
This work was supported by Nature Science Foundation of China (32160795 and U1802234), the Industrial Innovation Talent Project of the “Xing Dian Talent Support Program” of Yunnan Province in 2022 (XDYC-CYCX-2022-0029), the Young Talent Project of the “Xing Dian Talent Support Program” of Yunnan Province in 2023 (XDYC-QNRC-2023-0403), Yunnan Swine Industry Technology System Program (2023KJTX016), and the special Fund for Anhui Agriculture Research System (AGCYJSTX-05-15). Computational resources were supplied by Shanghai BIOZERON Biotechnology Co., Ltd. We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Author information
Authors and Affiliations
Contributions
H.H., X.B. and H.B.P. designed and conceived the study. F.Y.Y., L.R.M., J.J.Z., X.D. and L.M. collected the samples. Y.T., Q.L. and Y.F.L. performed data analysis. H.H., Y. H., X.B., K.P.W. and H.B.P. wrote and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, H., Huang, Y., Yang, F. et al. Metagenome-assembled microbial genomes (n = 3,448) of the oral microbiomes of Tibetan and Duroc pigs. Sci Data 12, 141 (2025). https://doi.org/10.1038/s41597-025-04413-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04413-1
