
Abstract
The Yadong trout (Salmo trutta), a species endemic to the Yatung River in Tibet, China, was classified as a second-class protected species in the 20th century. Now, it is considered one of the most important fishery resources in China. In this study, we assembled a near-complete genome of the S. trutta, integrating PacBio HiFi, Hi-C, and ONT sequencing technologies. The genome assembly spans 2.49 Gb, with 96.87% of the sequence anchored onto 40 chromosomes. In this assembly, a total of 12 chromosomes were assembled to a gap-free level, with 8 of them reaching the telomere-to-telomere level. The completeness of this assembly was assessed at 99.50% by BUSCO, containing approximately 63.24% repetitive sequences, and predicted to encode 41,782 protein-coding genes. This is the first near-complete genome assembly of the S. trutta, providing an essential resource for molecular breeding and germplasm conservation of this important species.
Similar content being viewed by others


Background & Summary
Salmo trutta, a member of the Salmonidae family, is characterized by a grey upper body with spots distributed above and below the lateral line1. It is native to Europe, Western Asia, and North Africa2, and can be broadly categorized into anadromous and lacustrine populations3. Since the mid-19th century, S. trutta has been introduced to 24 countries, including Russia, the United States, the United Kingdom, Japan, and countries in South America. Its strong migratory abilities and adaptability to diverse environments have enabled it to rapidly establish itself as a global species3,4.
In 1866, S. trutta was introduced to the Yadong River in Tibet, China, where it has since become a localized population, colloquially known as Yadong trout5. The specific environmental conditions of the Yadong River result in a slow reproductive and growth cycle for the Yadong trout population. This makes the species highly vulnerable to overfishing6,7. This has led to a steady decline in population numbers, prompting its designation as a second-class protected aquatic species in the Tibet Autonomous Region in 19928. In recent years, efforts by the Yellow Sea Fisheries Research Institute of the Chinese Academy of Fishery Sciences have led to the implementation of large-scale aquaculture programs to support the conservation and commercial farming of Yadong trout.
Advancements in sequencing technologies have made telomere-to-telomere (T2T) level genome assemblies feasible. As a result, several fish species, including Neosalanx taihuensis9, Lateolabrax maculatus10, and Clarias gariepinus11, have had their genomes published at this level of detail. However, the only available genome (GCA_901001165.1) of S. trutta remains the chromosomal-level assembly published by the Wellcome Sanger Institute in 201912, and the number of published chromosomal-level genomes is still extremely limited compared to the global distribution of different populations of S. trutta.
In this study, we integrated PacBio high-fidelity (HiFi), high-throughput chromatin conformation capture (Hi-C) and Oxford Nanopore Technologies (ONT) reads to assemble the S. trutta genome, achieving near complete genome sequence level (Fig. 1a,b). Compared to the published S. trutta genome12, our assembly shows significant improvements in continuity and completeness. This high-quality genome will provide valuable resources for the molecular breeding of S. trutta and facilitate comparative genomic analyses among S. trutta populations from different regions.

S. trutta genome snail plot and circos plot. (a) The snail plot presents the basic metrics of the genome assembly. (b) The circos plot represents the following metrics from outer to inner layers: (a) chromosomes, (b) gene density, (c) CG content, (d) DNA transposons, (e) LTRs, (f) LINEs, and (g) SINEs. The points on the chromosome backbone indicate detected telomeres, with red points representing chromosomes that are telomere-to-telomere with no gaps.).
Methods
Sample collection and sequencing
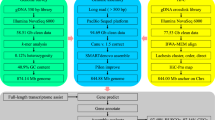
An adult female S. trutta was sourced from the Yadong County Industrial Park, Shang Yadong Township, Shigatse City, Tibet, China. We employed a rigorously annotated SDS method to obtain sufficient quality and quantity of genomic DNA (gDNA). The sheared gDNA was purified using AMPure PB beads, followed by end repair, adapter ligation, and further purification to construct SMRTbell templates. These templates were then loaded into SMRT cells and sequenced on the PacBio Sequel II platform, yielding 94.96 Gb (~38×) of HiFi data (Table 1).
For ONT data, DNA was extracted from fin clip tissue using the NEB Monarch® HMW DNA Extraction Kit for Tissue. Libraries were prepared and sequenced on the PromethION platform, resulting in 72.72 Gb (~29×) of ONT data (Table 1).
For Hi-C data, muscle tissue was processed through formaldehyde crosslinking, followed by washing, lysis, enzymatic digestion, DNA end modification, fragment ligation, purification, DNA end repair, biotin labeling, and PCR amplification. Illumina PE150 sequencing was performed, yielding 255.69 Gb (~103×) of Hi-C data (Table 1).
For WGS sequencing, DNA extracted from muscle tissues was transformed into Illumina library formats using the the NEBNext® Ultra™ DNA Library Prep Kit. Cluster formation for these libraries was carried out on cBot Cluster Generation System with the Illumina Paired-End Cluster Kit, adhering to the guidelines provided by the producer. We ultimately obtained 99.63 Gb (~40×) of short-fragment data (Table 1).
For RNA-seq data, we extracted RNA from nine different tissues, including muscle, liver, intestine, ovary, brain, spleen, gill, kidney, and heart. RNA-Seq libraries were assembled following the protocol of the library preparation kit. Illumina sequencing yielded an average of 6.83 Gb of data per tissue sample (Table 1). Additionally, we extracted RNA from a mixed tissue sample, and qualified RNA samples underwent reverse transcription, end repair, DNA fragmentation, adapter ligation, and amplification to construct the library. Sequencing was then carried out on the PacBio Sequel IIe platform, resulting in 10.11 Gb of Isoform Sequencing data. (Table 1).
Genome assembly and telomeres identification
To obtain a high-quality genome for S. trutta, we integrated ONT data with HiFi data and utilized Hifiasm13 (v0.19.9) to assemble a draft genome. Subsequently, we employed CRAQ14 (v1.0.9) to identify chimeric fragments and generated CRAQ-corrected genome. We then used kmerDedup15 for assembly redundancy removal and HapHic16 (v1.0.6) together with Hi-C data, to anchor the draft genome to 40 chromosomes, consistent with the chromosome number of the previously published S. trutta genome12 (Fig. 2a). We utilized Juicer-box17 (v1.91) for minor manual refinements of the genome, subsequently employing TGS-GapCloser18 (v1.2.1) in conjunction with ONT data to fill gaps within the genome. Thereafter, we leveraged NextPolish219 (v0.2.1), in tandem with HiFi and WGS data, to rectify the genome, culminating in the acquisition of a 2.49 Gb genome with a contig N50 of 47.99 Mb (Fig. 1a). We utilized the TeloExplorer parameter in quarTeT20 (v1.2.1) to identify the telomeres (TTAGGG) at both ends of each chromosome in the S. trutta genome, revealing that 18 chromosomes had double-end telomeres detected, 17 chromosomes had single-end telomeres detected, and 8 chromosomes achieved gap-free telomere-to-telomere status (Fig. 1b and Table 2).

Hi-C heatmap and collinearity dot plot of the genome. (a) The Hi-C heatmap illustrates the chromosome interaction frequencies of the S. trutta genome, with each blue contour representing a chromosome. (b) The dot plot displays the collinearity relationship with previously published genome assembly of S. trutta.
Additionally, we conducted a collinearity analysis using Minimap221 (v2.28) with the published S. trutta genome and visualized the results using pafCoordsDotPlotly.R (https://github.com/tpoorten/dotPlotly). The results showed that most chromosomes exhibited good collinearity, although some chromosomes displayed different arrangements (Fig. 2b). We also compared various assembly metrics between the two genomes, and found that our assembled genome demonstrated superior performance, with a total genome length of 2.49 Gb, an anchoring rate of 96.87%, a contig N50 of 47.99 Mb, a BUSCO completion rate of 99.50%, fewer genome gaps, 12 chromosomes being gap-free, and the detection of 53 telomeres (Table 3).
Repetitive sequence annotation
For the identification and characterization of genomic repetitive elements, we employed a combination of de novo prediction and homology-based annotation. We employed LTR-Finder22 (v1.07) to predict LTR retrotransposons in the S. trutta genome. Simultaneously, we utilized RepeatModeler23 (v2.0.5) to perform de novo predictions of repetitive elements, and based on the results from both methods, we constructed a repeat element database specific to the S. trutta genome with the Repbase database24 (v202101). Homology-based annotation was mainly conducted using the RepeatProteinMask and Repbase modules of RepeatMasker25 (v.4.1.0), also with default parameters for prediction. The predictions from both strategies were then integrated and filtered. The annotation results indicated that repetitive sequences account for approximately 63.24% of the genome, with the largest portion being occupied by various interspersed elements: long interspersed nuclear elements (LINEs) made up 32.34%, short interspersed nuclear elements (SINEs) constituted 0.55%, and long terminal repeats (LTRs) accounted for 10.80% (Fig. 1b and Table 4).
Genomic structure and functional annotation
Building upon the repetitive sequence-masked S. trutta genome, we employed Augustus26 (v3.5.0) for gene de novo prediction using default parameters. Additionally, we utilized Miniprot27 (v0.13) to perform homology annotation with protein sequences from four species, including Salmo salar, S. trutta, Oncorhynchus mykiss, and Oncorhynchus tshawytscha. Concurrently, we aligned RNA-seq data to the genome using HISAT228 (v2.2.1), followed by transcriptome assembly with StringTie29 (v2.2.3). Open reading frame (ORF) within the assembled transcripts were further identified using TransDecoder (https://github.com/TransDecoder/TransDecoder), and potential coding regions were further predicted to construct evidence for annotation. Additionally, we processed full-length transcriptome data using the IsoSeq330 (v4.0.0) pipeline and aligned them to the genome with GMAP (https://github.com/juliangehring/GMAP-GSNAP) to generate annotation evidence. Finally, we integrated four annotation strategies—de novo, homology, transcript-based, and full-length transcriptome—using EvidenceModeler31 (v1.1.1). The final gene set was further refined by aligning it to the S. trutta genome annotation using Liftoff32 (v1.6.3) as a reference, resulting in the identification of 41,782 genes. To assess the accuracy of gene annotation, we compared the distribution of mRNA lengths and exon counts per mRNA in the S. trutta genome with gene data from S. salar and O. mykiss. The results demonstrated highly similar genomic component distribution characteristics among the three species (Fig. 3a).

Comparative genome plot of closely related species and gene functional annotation upset plot. (a) Comparison of mRNA length distribution and exon count per mRNA among the three closely related species. (b) Upset plot of gene functional annotation using data from EggNOG, Pfam, KEGG, NR, Kofam, and SwissProt.
The predicted gene protein sequences were aligned against various functional databases using Diamond33 (v2.1.6) with an E-value threshold set at 1e-5. The databases encompassed Kyoto Encyclopedia of Genes and Genomes34 (KEGG), Swiss Institute of Bioinformatics Protein Database35 (Swiss-Prot), Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups36 (EggNOG), Protein Families Database37 (Pfam), KOfam Database38 (Kofam), and the Non-Redundant database39 (NR) to extract potential gene functional information, which was subsequently utilized for statistical analysis. A total of 41,629 genes, which account for 99.63% of the total estimated protein-coding genes, have been effectively annotated by a minimum of one of these databases (Fig. 3b and Table 5).
Data Records
Technical Validation
We employed BUSCO43 (v5.3) with the Actinopterygii database (actinopterygii_odb10) to evaluate the genome assembly’s completeness. The BUSCO analysis of the genome revealed an overall completeness of 99.50%, with 56.48% being single-copy and 43.02% being duplicated, leaving only 0.28% as fragmented and 0.22% missing (Fig. 1a and Table 3). We utilized Inspector44 (v1.0.1) to map the PacBio HiFi reads against the genome, achieving a quality score, as indicated by the Quality Value (QV), of 44.91 and a read-to-contig alignment rate of 99.95%. Additionally, we applied CRAQ14 for genome assessment, which determined an Assembly Quality Index (AQI) of 96 (>90), indicating that our assembled genome is highly complete, of good quality, and has achieved a reference-quality standard.
Code availability
In this study, we did not employ any customized scripts or software for personalized analysis. The analytical tools and parameters used are described in the methods section. For software without specific parameter descriptions, the default parameters were selected.
References
Moyle, P. B. Inland Fishes of California. (University of California Press, 1976).
Lobry, J., Mourand, L., Rochard, E. & Elie, P. Structure of the Gironde estuarine fish assemblages: a comparison of European estuaries perspective. Aquat. Living Resour. 16, 47–58 (2003).
Klemetsen, A. et al. Atlantic salmon Salmo salar L., brown trout Salmo trutta L. and Arctic charr Salvelinus alpinus (L.): a review of aspects of their life histories. Ecol Freshwa Fish 12, 1–59 (2003).
Elliott, J. M. Quantitative ecology and the brown trout Ch. 9 (Oxford Univ. Press, 1994).
Liu, J. et al. Comparative Analysis of Nutrient Composition of Different-Colored Yadong Trout Eggs. Prog Fish Sci 44, 133–141 (2023).
Kang, B. et al. Introduction of non-native fish for aquaculture in China: A systematic review. RevAquac 15, 676–703 (2023).
Zhou, J., Min, Z., Zhang, C., Li, B. & Wang, W. Research progress of fishery resources in Tibet. Anim. Husb. Feed Sci. 8, 246–250 (2016).
Tian, H.-F., Hu, Q.-M. & Li, Z. Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers in swamp eel (Monopterus albus). Sci Prog 104, 368504211035597 (2021).
Zhou, Y. et al. Gap-free genome assembly of Salangid icefish Neosalanx taihuensis. Sci Data 10, 768 (2023).
Sun, Z. et al. Telomere-to-telomere gapless genome assembly of the Chinese sea bass (Lateolabrax maculatus). Sci Data 11, 175 (2024).
Nguinkal, J. A. et al. Haplotype-resolved and near-T2T genome assembly of the African catfish (Clarias gariepinus). Sci. Data 11, 1095 (2024).
Hansen, T. et al. The genome sequence of the brown trout, Salmo trutta Linnaeus 1758. Wellcome Open Res 6, 108 (2021).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat. Commun. 14, 6556 (2023).
Abalde, S., Tellgren-Roth, C., Heintz, J., Vinnere Pettersson, O. & Jondelius, U. The draft genome of the microscopic Nemertoderma westbladi sheds light on the evolution of Acoelomorpha genomes. Front. Genet. 14, (2023).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat. Plants 10, 1184–1200 (2024).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98 (2016).
Xu, M. et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9, giaa094 (2020).
Hu, J. et al. NextPolish2: A Repeat-aware Polishing Tool for Genomes Assembled Using HiFi Long Reads. Genomics Proteomics Bioinf 22, qzad009 (2024).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, uhad127 (2023).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.1–4.10.14 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Guizard, S. et al. nf-core/isoseq: simple gene and isoform annotation with PacBio Iso-Seq long-read sequencing. Bioinformatics 39, btad150 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
A, S. & Sl, S. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, (2021).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18, 366–368 (2021).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 45–48 (2000).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–D314 (2019).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–D419 (2021).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33, D501–504 (2005).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_043791845.1 (2024).
Li, C. Salmo trutta genome. Figshare. https://doi.org/10.6084/m9.figshare.27282591.v3 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP540059 (2024).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654 (2021).
Chen, Y., Zhang, Y., Wang, A. Y., Gao, M. & Chong, Z. Accurate long-read de novo assembly evaluation with Inspector. Genome Biology 22, 312 (2021).
Acknowledgements
This research was funded by the National Key R&D Program of China (2024YFD2400901 and 2022YFD2400100), the Key Research and Development Project of Shandong Province (2024LZGC005),the AoShan Talents Cultivation Program Supported by Qingdao National Laboratory for Marine Science and Technology (grant number 2017ASTCP-ES06), the Taishan Scholars Program (NO. tstp20221149) to C.S, the National Ten-Thousands Talents Special Support Program to C.S, the Key R&D Program of Hebei Province, China (21326307D) and the Central Public-interest Scientific Institution Basal Research Fund, CAFS (grant number 2023TD19).
Author information
Authors and Affiliations
Contributions
C.S. conceived of the project. C.L. and S.H. analyzed the data. C.L. drafted the manuscript. S.L. K.L., H.W. and Q.W. revised the manuscript. Y.L. and C.L. prepared the sample materials and extracted the DNA. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, C., Han, S., Li, S. et al. Near complete genome assembly of Yadong trout (Salmo trutta). Sci Data 12, 74 (2025). https://doi.org/10.1038/s41597-025-04418-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04418-w
