
Abstract
Black carp (Mylopharyngodon piceus) is one of the “four famous domestic fishes” in China and an important economic fish in freshwater aquaculture. A high-quality genome is essential for advancing future biological research and breeding programs for this species. In this study, we aimed to generate a high-quality chromosome-level genome assembly of black carp using Nanopore and Hi-C technologies. The final genome assembly was 848.70 Mb in length, with a contig N50 of 3.37 Mb and a scaffold N50 of 34.13 Mb. The genome was anchored onto 24 chromosomes by Hi-C technology. The BUSCO (Benchmarking Universal Single-Copy Orthologs) completeness score of the genome assembly is 97.6% when compared to the Actinopterygii_odb10 database. In total, 203.54 Mb of repeat sequences and 37,418 protein-coding genes were predicted in the genome assembly. Taken together, our study provides a chromosome-level genome assembly, which can serve as a valuable genetic resource to support further biological studies and breeding efforts of black carp.
Similar content being viewed by others
Background & Summary
The black carp (Mylopharyngodon piceus, NCBI Taxonomy ID: 75356) belongs to the genus Mylopharyngodon within the family Xenocyprididae and order Cypriniformes1. This demersal fish is native to East and Southeast Asia, predominantly distributed in amur river basin, as well as various rivers and lakes across southern China and Vietnam1. It has also been introduced to numerous countries in North America, Europe, Africa, and the Middle East2.
Recognized as one of the largest cyprinids in the world, the black carp can reach a maximum body length of 1.5 meters and a body weight of 70 kilograms3. Renowned for its tasty meat, high nutritional value, simple aquaculture process, and fast growth rate, black carp is one of the most traditionally economic fish in China, where it has been cultivated and consumed since the Tsin Dynasty (A.D. 265–420)4,5. Currently, black carp remains one of the most dominant freshwater farmed fish in China, commonly referred to as one of the culturally significant “four famous domestic fishes” along with the grass carp (Ctenopharyngodon idella), silver carp (Hypophthalmichthys molitrix), and bighead carp (Hypophthalmichthys nobilis)5. According to the China Fishery Statistical Yearbook, black carp farming production in China reached 748,026 tons in 2022, marking an increase of more than 50% from a decade earlier4,6.
Beyond its culinary appeal, black carp also plays a role in biological control within aquaculture. As a carnivorous fish that feeds primarily on snails, it has been introduced to many countries, including the United States, for the management of snail populations in aquaculture facilities that can otherwise impact aquaculture operations3,7.
The large-scale application of sequencing technologies since the 21st century has facilitated the availability of whole-genome sequences for around 1400 fish species in NCBI databases8. The wealth of genomic information has been instrumental in reconstructing the evolutionary history of fish and advancing basic research in areas such as disease resistance, immune responses and morphogenesis9,10,11. These genomic insights also support conservation and aquaculture practices. The first draft genome of black carp derived from short-read sequencing was reported in 202212, and it was optimized in 202313, with a contig number reduced to 3,436 and a contig N50 enhanced to 2.9 Mb. However, both of the published assemblies remain at the scaffold level, which constrains the understanding and utilization of valuable genomic information for this species. Recently, a chromosome-level genome of black carp assembled from PacBio HiFi reads was reported14, with a genome size of 893.89 Mb and a scaffold N50 of 36.19 Mb.
In this study, we present a high-quality chromosome-anchored genome assembly of black carp from a different source. We combined our Nanopore long reads with published Illumina short reads12,15 to generate a genome assembly of black carp, followed by Hi-C sequencing to scaffold the assembled sequences to chromosomes. The resulting genome spanned 848.70 Mb, with a contig N50 of 3.37 Mb and a scaffold N50 of 34.13 Mb, and was anchored onto 24 chromosomes. Compared to the previously reported chromosome-level genome of black carp, our genome assembly exhibits a size difference of over 45 Mb, reflecting genetic diversity between populations. Furthermore, our assembly showed high completeness, with a BUSCO score of 97.6%. A total of 23.98% (203.54 Mb) of the genome was identified as repeat sequences, and 37,418 protein-coding genes were predicted. Our chromosome-level genome offers critical genomic data for black carp, generated using a distinct methodology and representing a different genetic population. It provides an alternative but equally valuable reference genome to advance breeding programs and biological studies for this species. Additionally, our assembly enriches the genomic resource pool of black carp, enabling further investigations into genome evolution, local adaptation, and genetic diversity through comparative genomics.
Methods
Sample collection and genome sequencing
One healthy black carp with a body length of 23.2 cm (sex not determined), collected from Jingyang Hatchery in Guangdong Province, China, was sampled for Nanopore and Hi-C sequencing. After sampling, the muscle tissues of these fish were first frozen in liquid nitrogen and subsequently delivered to a refrigerator at -80°C for storage until sequencing began.
For Nanopore sequencing, the genomic DNA of muscle tissue was extracted using the Monarch Genomic DNA Purification kit (NEB, #T3010), following the standard protocol. The genomic DNA was assessed in terms of quantity, purity, and integrity, using NanoDrop Spectrophotometer (Thermo Fisher Scientific) and 1.5% agarose gel electrophoresis. The Nanopore libraries were constructed from 4 µg of high-quality genomic DNA by utilizing the LSK-110 (ONT, Oxford, United Kingdom) library preparation kit. Long-read sequencing was performed on the in-house MinION (ONT) platform using R.9.4.1 Flow Cell (FLO-MIN106, ONT). The resulted fast5 files were base-called with Guppy v6.0.1 (https://community.nanoporetech.com/docs/prepare/library_prep_protocols/Guppy-protocol/v/gpb_2003_v1_revax_14dec2018) in the “sup” accurate model. After the removal of adapters, 35.83 Gb of Nanopore sequencing reads were obtained, with an average read length of 3,293 bp and an N50 read length of 7,485 bp (Table 1). This equates to a 42x coverage of the genome based on the size of our final genome assembly.
To achieve the chromosome-level genome assembly of black carp, Hi-C sequencing was performed using DNA from the muscle tissue. All steps, from sample preparation to DNA extraction, were performed in-house following the restriction enzyme Pore-C (RE-Pore-C) protocol16 provided by Oxford Nanopore Technologies. The prepared DNA libraries were then sequenced by Illumina platform (Illumina Inc., San Diego, CA, USA) at Novogene Co., Ltd. (Hong Kong, China) using the 150 bp paired-end (PE) mode, which yielded 53.25 Gb of raw sequencing data (Table 1).
Genomic feature survey
The genome size and heterozygosity of black carp were analysed using the available Illumina sequencing data (NCBI SRA ID SRR14181237)12,15. The Illumina short reads served as input for Jellyfish v2.3.017, from which a k-mer (k = 21) frequency distribution was obtained, as depicted in Fig. 1. After discarding the k-mers with abnormal depth, the genome size was estimated by using the formula genome size = k-mers number/average depth of k-mers. Consequently, the estimated genome size of black carp was 765.00 Mb, with a heterozygosity rate of 0.355%.

k-mer (k = 21) frequency distribution generated using Illumina sequencing data of black carp. The x-axis and y-axis represent the depth and frequency of the k-mer.
De novo genome assembly and evaluation
The de nove genome assembly of black carp was performed by combining the Nanopore and Illumina data. Briefly, the Nanopore sequencing reads were used for genome assembly by NextDenovo v2.5.0 (https://github.com/Nextomics/NextDenovo) with default parameters. Then, to enhance the accuracy of the assembly, NextPolish v1.4.118 was employed for polishing with Nanopore long reads and published Illumina short reads12,15. This step generated an assembly of 848.42 Mb, containing 721 contigs, with a contig N50 of 3.37 Mb.
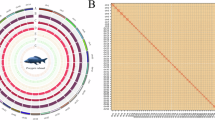
For Hi-C reads, initial filtering was conducted to remove those of low quality by using Trimmomatic v0.3919 with the parameters of “ILLUMINACLIP:NexteraPE-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36”. Subsequently, Juicer v1.620 was used to find the potential linkage between contigs and scaffolds within the assembly. 3D-DNA phasing branch 20100821 was used to anchor the assembled sequences onto chromosomes, leading to the generation of a contact map that was visualized using Juicebox v1.11.0822. Finally, we obtained a chromosome-level genome assembly of 848.70 Mb, which was scaffolded by Hi-C technique onto 24 pseudo-chromosomes with sizes ranging from 20.78 Mb to 48.76 Mb (Table 2 and Fig. 2). The assembly comprises 172 scaffolds in total, with a scaffold N50 of 34.13 Mb.

Characteristics of the black carp genome. (a) Hi-C contact map of the black carp genome assembly. (b) Circos plot of the black carp genome. From the inner to the outer layers: gene density, transposable element (TE) density, GC content, and chromosome ideograms.
To evaluate the completeness of the assembled genome, we employed BUSCO (Benchmarking Universal Single-Copy Orthologs) v5.1.223, utilizing 3,640 orthologs from the Actinopterygii_odb10 database as reference. The BUSCO analysis revealed 3,552 complete Benchmarking Universal Single-Copy Orthologs within our assembly, indicating a 97.6% completeness level (Table 2). We also mapped the published Illumina short reads of black carp to the assembled genome by BWA v0.7.1824 to assess its consistency. Analysis of the mapping results using SAMtools v1.2025 confirmed that 97.97% of the Illumina reads were successfully mapped to the genome, with a coverage of 99.53%.
Repetitive sequence annotation
For the prediction of repeats in our black carp genome assembly, we integrated both homology-based and de novo methodologies. First, RepeatModeler v2.0.326 was employed for the de novo discovery of repetitive sequences, creating a custom library tailored to our genome. Subsequently, a comprehensive library was constructed by merging sequences from Dfam v3.627 and Repbase28 with those identified by RepeatModeler. With this enriched library as a reference, we utilized RepeatMasker v4.1.329 to detect the repetitive sequences throughout the genome. This approach led to the identification of approximately 203.54 Mb of repeats, which constitute 23.98% of the total genome assembly (Table 3).
Protein-coding gene annotation
The pipelines based on ab initio prediction, transcript-assisted strategies, and protein alignment were used to predict protein-coding genes. Seventeen RNA-Seq datasets from various tissues, including hindgut, foregut, eye, fin, gill, head, kidney, liver, muscle, bladder, brain, skin, and spleen, were retrieved from the NCBI repository30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46 for the prediction. Firstly, BRAKER v3.0.847 was employed for an ab initio gene structure prediction, utilizing the transcript evidence derived from the alignment of RNA-seq reads by HISAT v2.2.148 against the repeat-masked genome assembly. The second pipeline involved both de novo and genome-guided transcriptome assembly strategies by Trinity v2.15.149 prior to protein-coding gene prediction. Then the two versions of transcriptome assemblies were combined and the gene structures were predicted by using the PASA pipeline v2.5.350. For protein alignment-based prediction, the protein sequences from the UniProt (https://www.uniprot.org) were input to miniport v0.13-r262-dirty51 with default parameters to generate the protein-base file. Ultimately, EVidenceModeler v2.0.052 was used to produce the final gene model by merging the output resulting from PASA, BRAKER, and miniprot with the weights in Table 4. Overall, our approach, combining the ab initio prediction, transcript-based strategies, and protein alignment, predicted a total of 37,418 protein-coding genes within our genome assembly. These genes exhibited an average of 7.78 exons and an average exon length of 160 base pairs (Table 5). The BUSCO score for the predicted genes is 91.2%, indicating a high level of completeness in the predictions. For gene functional annotation, we used eggNOG-mapper v2.1.1253 and InterProScan v5.69-101.054 for ortholog- and protein domain-based annotations, which yielded 24,734 (66.1%) and 29,667 (79.3%) hits, respectively. By integrating the results from both annotation methods, a total of 30,278 (80.9%) genes were successfully assigned with functional annotations.
Data Records
The sequencing data and genome assembly have been submitted to the public repositories. The Hi-C and Nanopore sequencing data have been deposited in the NCBI Sequence Read Archive database under the SRA accession # SRR2876216455 and SRR2876216556, and BioProject accession # PRJNA1102922. The genome assembly has been deposited at the NCBI GenBank under the accession # JBCHWC00000000057.
Technical Validation
To evaluate the quality of our assembly, we used BUSCO v5.1.223 to assess its completeness. The BUSCO results confirmed that 3,552 (97.6%) of the 3,640 conserved single-copy genes in Actinopterygii are in our assembled genome, implying a high degree of completeness. Additionally, to evaluate the accuracy of our assembly, we mapped the Illumina short reads to the genome using BWA v0.7.1824. Based on the statistical analysis performed with SAMtools v1.2025, 97.97% of the Illumina short reads were mapped to the genome and the coverage was 99.53%, which indicated a high level of accuracy. Collectively, these results demonstrated that our assembly is of high quality.
Code availability
All commands used in this study were executed according to the manuals and protocols of the corresponding software. No specific scripts were used in this study.
References
FishBase. Mylopharyngodon piceus https://fishbase.mnhn.fr/summary/SpeciesSummary.php?ID=4602&AT=black+carp (2023).
Siriwardena, S. Mylopharyngodon piceus (black amur) https://www.cabidigitallibrary.org/doi/10.1079/cabicompendium.73511 (2019).
Nico, L. G., Williams, J. D. & Jelks, H. L. Black Carp: Biological Synopsis and Risk Assessment of an Introduced Fish. (American Fisheries Society Special Publication, 2005).
Chen, P. & Arratia, G. Oldest known Mylopharyngodon (Teleostei: Cyprinidae) from the Mongolian Plateau and its biogeographical implications based on ecological niche modeling. Journal of Vertebrate Paleontology 30, 333–340, https://doi.org/10.1080/02724631003620930 (2010).
Tang, H., Mao, S., Xu, X., Li, J. & Shen, Y. Genetic diversity analysis of different geographic populations of black carp (Mylopharyngodon piceus) based on whole genome SNP markers. Aquaculture 582, 740542, https://doi.org/10.1016/j.aquaculture.2024.740542 (2024).
Chinese Ministry of Agriculture. China Fishery Statistical Yearbook. (China Agriculture Press, 2023).
Whitledge, G. W. et al. Establishment of invasive Black Carp (Mylopharyngodon piceus) in the Mississippi River basin: identifying sources and year classes contributing to recruitment. Biological Invasions 24, 3885–3904, https://doi.org/10.1007/s10530-022-02889-1 (2022).
National Center for Biotechnology Information. Genome https://www.ncbi.nlm.nih.gov/genome/?term=fish (2024).
Liu, Z. et al. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nature Communications 7, 11757, https://doi.org/10.1038/ncomms11757 (2016).
Lu, G. & Luo, M. Genomes of major fishes in world fisheries and aquaculture: Status, application and perspective. Aquaculture and Fisheries 5, 163–173, https://doi.org/10.1016/j.aaf.2020.05.004 (2020).
Lu, Y. et al. A chromosome-level genome assembly of the jade perch (Scortum barcoo). Scientific Data 9, 408, https://doi.org/10.1038/s41597-022-01523-y (2022).
Lu, Y. et al. Genome survey sequence of black carp provides insights into development-related gene duplications. Journal of the World Aquaculture Society 53, 1197–1214, https://doi.org/10.1111/jwas.12870 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc:JAQQBF000000000.1 (2023).
Wang, C. et al. Genomic features for adaptation and evolutionary dynamics of four major Asian domestic carps. Science China-Life Sciences 67, 1308–1310, https://doi.org/10.1007/s11427-023-2479-2 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR14181237 (2021).
Oxford Nanopore Technologies. Restriction Enzyme Pore-C https://community.nanoporetech.com/extraction_methods/restriction-pore-c (2020).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2019).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Research 41, D70–D82, https://doi.org/10.1093/nar/gks1265 (2012).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research 110, 462–467, https://doi.org/10.1159/000084979 (2005).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 25, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR10357871 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR10357872 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR10357873 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR10357874 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR10357875 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR10357876 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702070 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702071 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702072 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702073 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702074 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702075 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702076 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702077 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702078 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702079 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR23702080 (2023).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiv https://doi.org/10.1101/2023.06.10.544449 (2024).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, https://doi.org/10.1093/bioinformatics/btad014 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular Biology and Evolution 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28762164 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28762165 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBCHWC000000000 (2024).
Acknowledgements
This study was supported by the APRC-CityU New Research Initiatives/Infrastructure Support from Central (9610574) to WC. This work was supported by the Hetao Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone Shenzhen Park Project (HZQB-KCZYZ-2021017); Early career scheme (project number CityU 21100521) from the Hong Kong Research Grant Council; and new Research Initiatives support from City University of Hong Kong (project number 9610497) to R.L.
Author information
Authors and Affiliations
Contributions
R.L. and W.C. conceived and supervised this work. L.Z., J.Z. and W.Z. collected the samples and assisted with data analysis. Y.S. prepared DNA for Nanopore and Hi-C sequencing and performed the Nanopore sequencing. X.L. and Z.Z. conducted the bioinformatics analysis. Y.Z. conducted the data analysis and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Shao, Y., Liu, X. et al. Chromosome-level genome assembly of black carp Mylopharyngodon piceus using Nanopore and Hi-C technologies. Sci Data 12, 145 (2025). https://doi.org/10.1038/s41597-025-04421-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04421-1
