
Abstract
This contribution presents a comprehensive extension of the QM9 dataset (originally at 133 K molecules) with the calculation of G4MP2 enthalpies for 9,841 molecules, featuring up to nine heavy atoms. We present QM9-LOHC, a (de)hydrogenation dataset of 10,373 reactions, including a minimum of 5.5% weight hydrogen storage capacity in line with the Department of Energy standards for Liquid Organic Hydrogen Carriers (LOHC). By utilizing the accurate quantum chemical method G4MP2 we expand the QM9 database and explore new avenues for the exploration of hydrogen storage technologies (electrochemical LOHCs, alkali metal-LOHCs, and mixtures of LOHCs). The QM9-LOHC dataset, with its focus on reactions that vary only by hydrogen saturation levels, provides a needed data resource for advancing the design and optimization of both conventional and innovative LOHC systems, and high-fidelity data for molecular discovery.
Similar content being viewed by others
Background & Summary
The quest for sustainable, efficient, and carbon-free energy storage solutions has brought hydrogenation reactions to the forefront of scientific research1,2,3,4,5,6. Hydrogen, with its high energy density and clean combustion, is a promising candidate for future energy systems3,7,8,9,10. However, its storage and transport pose significant challenges due to its low volumetric energy density and the complexities of handling gaseous substances11,12,13. Thus, reactions involving the chemical storage and release of hydrogen, particularly those involved in Liquid Organic Hydrogen Carriers (LOHCs), emerge as vital components in efficient and safe hydrogen storage3,8,14,15,16,17,18. LOHC systems offer a promising approach to the chemical storage of hydrogen, thereby addressing the limitations of high pressure hydrogen-storage methods14,19,20.
LOHCs operate by absorbing hydrogen atoms via hydrogenation reactions (add hydrogens to the unsaturated bonds) and releasing them via dehydrogenation reactions3,18. The efficacy of the technology depends on the identification of organic carrier molecules that can reversibly store hydrogen in high densities while being economically viable, catalytically stable, and environmentally benign17,19. Therefore, the study of hydrogenation reactions, specifically focusing on their thermodynamic and kinetic properties, is crucial for advancing LOHC technology3,14. Within the scope of LOHC research, several chemical systems have garnered particular interest due to their unique characteristics and potential applications, four categories of which are shown in Fig. 1 and described below:
-
a.
Conventional LOHC Systems: These systems employ a catalyst-mediated hydrogenation/dehydrogenation cycle, such as benzene/cyclohexane, toluene/methyl-cyclohexane, and N-ethylcarbazole(NEC)/dodecahydro-N-ethylcarbazole. Central to their operation is the catalytic efficiency and the ability to store hydrogen reversibly without significant degradation of the LOHC molecule. Representing one of the first approaches to chemical storage of hydrogen in liquid carriers, these conventional systems set the stage for modern LOHC technology, offering a proven, viable pathway for sustainable energy integration and marking a significant stride towards a cleaner, hydrogen-based economy14,18,21,22,23,24,25.
-
b.
Mixture of LOHCs: By controlling the mole-ratios of different LOHC components, it is possible to tailor the physical properties of the system, such as its melting and boiling points. Such precise control enables the formulation of LOHC mixtures that remain liquid at ambient conditions, which is crucial for practical storage and transportation. Additionally, this method allows for the optimization of the system’s hydrogen storage capacity and thermal stability, while also enhancing the economic viability of the hydrogen carriers. This tailored approach promises to address both efficiency and cost concerns, paving the way for more adaptable and user-friendly hydrogen storage solutions3,26. Illustratively, Stark et al. reported a mixture 42% N-ethyl carbazole (NEC) and 58% N-propyl carbazole (NPC), originally melting at 342 K and 320 K respectively, lowers the mixture’s melting point to 297 K26.
-
c.
Electrochemical LOHCs: These systems are distinguished due to their capability for electrochemical reversibility, enabling them to be employed effectively for hydrogen storage and release. An illustrative example of such a system is the isopropanol/acetone pair, where electrochemical reactions are employed to either liberate or absorb hydrogen27. This feature can notably improve the efficiency and control of the hydrogenation and dehydrogenation processes, offering an alternative approach to LOHC technology27,28,29.
-
d.
Alkali Metal-LOHCs: These systems feature a substitution whereby an alkali metal replaces a proton in the carrier molecule, exemplified by systems such as Na-Phenoxide/Na-Cyclohexanolate30. This critical modification can considerably decrease the enthalpy of hydrogenation and dehydrogenation reactions, which may result in more efficient storage cycles and lower energy demands for releasing hydrogen. The potential for such systems to lower the overall reaction enthalpy of hydrogen storage makes them a compelling area of research within the field of LOHCs30,31,32.

Four categories of Liquid Organic Hydrogen Carrier (LOHC) molecules, as described in the text: Conventional, Electrochemical, Alkali Metal, and Mixtures of LOHCs. MP stands for melting point, ∆H is dehydrogenation enthalpy.
These categories and diverse approaches to LOHC technology reflect the breadth of research aimed at overcoming the challenges of hydrogen storage. Each category offers unique advantages and research opportunities for developing and deploying efficient, sustainable, and cost-effective hydrogen storage solutions.
Data-driven approaches have had a transformative impact in chemistry33,34,35, enabling, for example, innovation in molecular synthesizability predictions36,37,38,39, energy storage applications40,41,42,43, and drug discovery44,45,46. Accurate molecular data is essential for these machine learning efforts. One popular small molecule dataset is QM933,34, which provides, for 133 K molecules with 9 (or fewer) heavy (non-hydrogen) atoms (the GDB-9 subset of the larger GDB-1747 collection of molecules with 17 or fewer heavy atoms), properties computed using the B3LYP48 method and later refined49 with the more accurate composite quantum chemical method G4MP250,51,52,53.
The QM9 dataset contains computed properties (e.g.: optimum molecular geometries, enthalpies of formation, dipole moments, partial charges, and vibrational frequencies) for an extensive array of small organic molecules48,49. This invaluable dataset has facilitated the exploration and discovery of novel compounds and reaction pathways through computational methods54,55,56.
Recently we developed an in-silico discovery pipeline that employs cheminformatics and quantum chemical calculations to identify novel conventional LOHC molecules19. Using this pipeline, we screened the large GDB-17 database, containing 166 billion molecules, and the ZINC1557 database, containing 1.2 billion molecules, implementing a selection protocol that integrated machine learning to predict physical properties (melting/boiling points) and synthetic accessibility. Our efforts identified 41 novel LOHC molecules including benzofuran, benzoxazole groups, substituted quinolines and phenyl pyridines, all of which exhibit promising chemical properties for LOHC applications. In another study, Paragian et al. harnessed the QM9 database to predict hydrogenation enthalpies for over a million potential LOHC molecules from PubChem, identifying 37 prime candidates using machine learning models54. The success of both studies demonstrates that extensive datasets (e.g. GDB-1747, ZINC1557, QM948,49, and PubChem58) lay the groundwork to discover novel small molecules suitable for practical LOHC systems when they are effectively utilized33,34,35,59,60.
In this investigation, we compile a dataset of 10,373 (de)hydrogenation reactions derived from the QM9 dataset. Our selection process focused on chemical reactions that met two primary criteria: (a) the reactant and product pairs differ only by their levels of hydrogen saturation, and (b) the pairs exhibit a significant hydrogen storage capacity, specifically 5.5% by weight hydrogen or more, as set by the standards of the Department of Energy61. While additional constraints—such as an enthalpy of dehydrogenation (∆H) between 40 and 70 kJ/mol per H2 and being liquid at room temperature, along with safety, non-toxicity, stability, and reversibility—are essential for practical applications3,18,19, it is noteworthy that several of these properties can be tuned3,14,19,26,31. This tunability is possible by shifting from conventional LOHC technology to more innovative approaches (as described in the introduction, see Fig. 1). Such versatility in a technological approach broadens the scope of potential applications, accommodating a wider range of operational requirements.
To accurately determine the structures and enthalpies of dehydrogenation reactions, we performed 9,841 new quantum chemical calculations using the G4MP2 method. The 9,841 new quantum chemical calculations we performed were specifically aimed at determining the enthalpies of hydrogenated forms of unsaturated molecules, which, while already present in the original QM9 dataset, lacked their hydrogenated counterparts. This effort, combined with existing data from the original QM9 dataset, culminated in the creation of the QM9-LOHC62 open-access dataset, which encompasses 10,373 dehydrogenation reactions. We propose the QM9-LOHC dataset as a reference dataset for hydrogen storage technologies using LOHCs.
The QM9-LOHC dataset, with its newly calculated G4MP2 energies, extends the utility of the original QM9 database for in-depth studies of hydrogenation reactions, which are crucial in the development of advanced energy storage applications. Furthermore, the database will serve as a vital asset for the application of machine learning techniques in quantum chemistry, fostering the development of innovative methods that can accurately predict reaction enthalpies, particularly catering to the needs of energy storage research. This augmented database, moreover, is a significant step toward enabling the accurate calculation of reaction energies and provides a robust foundation for the development of predictive machine learning models for molecular discovery.
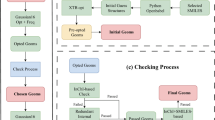
As shown in Fig. 2, the search for LOHC candidates started within the QM9 database47,48,49. First, We used the RDKit63 library to analyze the SMILES representations of molecules in the QM9 dataset, allowing us to identify those with unsaturated sites such as double or triple bonds, or unsaturated aromatic rings. Second, we deduced the saturated counterpart of each SMILES string from step 1 by simple string manipulation of unsaturated molecules. These two steps allowed us to identify over 100,160 dehydrogenation reactions of organic molecules, with each reaction consisting of a pair of molecules: a hydrogen-lean molecule and its hydrogen-rich counterpart. Third, we down selected by considering only reactions that have a hydrogen storage capacity of at least 5.5% wt. H2, as calculated with Eq. (1):
where MWH-rich and MWH-lean are the molar weights of the hydrogen-rich and the hydrogen-lean species, respectively. From this, we obtain a dataset of 10,373 reactions of pairs of unsaturated and saturated organic molecules.

Schematic of the protocol for developing the QM9-LOHC Dataset from the GDB-9 and QM9 databases49. The dataset includes 10,373 reactions with a minimum hydrogen storage capacity of 5.5% wt. H2, derived from selecting unsaturated molecules and generating their corresponding saturated forms. Representative molecules from the dataset are shown.
Upon the attempt to obtain G4MP2 enthalpy data from the QM9 dataset for the hydrogen-rich molecules, we identified that out of 10,373 molecules, only 532 were present. To fill in the data for the missing 9,841 molecular enthalpies, we perform quantum chemical calculations using Gaussian 16 software64 with the G4MP2 method50,51. G4MP2 is a composite method based on G4 theory that uses MP2 perturbation theory to obtain higher computational efficiency. A previous assessment of G4MP2 energies on a subset of QM9 molecules reported an accuracy of 0.79 kcal/mol (3.3 kJ/mol) with respect to accurate experimental enthalpies of formation49, showing that the G4MP2 method is highly accurate and reliable for molecules that are in or similar to the QM9 dataset.
Before running the quantum chemical calculations, the minimum energy conformers were obtained using the Universal Force Field (UFF) method in RDKit. The G4MP2 method employs B3LYP/6–31 G(2df,p) optimized geometries for a series of single-point energy calculations at higher levels of theory. The zero-point energy (E(ZPE)) is computed using B3LYP/6–31 G(2df,p) frequencies, which are scaled by a factor of 0.9854 to account for anharmonic effects. The nature of each located potential energy surface stationary point was confirmed as a minimum by the absence of imaginary frequencies. The initial energy calculation is performed at the coupled-cluster level of theory, CCSD(T), with the 6–31 G(d) basis set. This energy is subsequently refined by applying a series of corrections, including those derived from MP2 and Hartree-Fock (HF) energies and high-level corrections. This multi-step approach allows for the calculation of highly accurate total energies, benefiting from the computational efficiency of MP2 and the accuracy of coupled-cluster methods.
From the computed G4MP2 energies, the reaction enthalpies (ΔHrxn) of the 10,373 pairs are calculated using Eq. (2)
where H° is the absolute enthalpy (at 298.15 K and 1 atm) and nH2 is the number of moles of H2 involved in the reaction. For all what follows, ∆H refers to the standard gas-phase enthalpy of dehydrogenation reaction and is reported in units of kJ/mol H2 unless stated otherwise.
Data Records
The dataset is accessible via Zenodo at the following link62: https://doi.org/10.5281/zenodo.10926772. Contained within a zip file, the dataset comprises two CSV files: “QM9_G4MP2_all.csv” and “QM9-LOHC_new_molecules.csv”. The “QM9_G4MP2_all.csv” file encapsulates the entirety of the QM9-LOHC dataset, delineating 10,373 reactions. The data columns present include unsaturated SMILES strings (unsat_SMILE), saturated SMILES strings (sat_SMILE), dehydrogenation enthalpy measured in kJ/mol H2 (delta_H), the number of H2 molecules (nH2) involved in the dehydrogenation reaction, and the hydrogen storage capacity, %wt. H2 (pH2). The “QM9-LOHC_new_molecules.csv” file narrows its focus to a selection of the QM9-LOHC dataset, spotlighting saturated SMILES strings that represent novel molecules not identified in the original dataset. This file mirrors the columns found in the first, providing data on unsaturated SMILES strings (unsat_SMILE), saturated SMILES strings (sat_SMILE), dehydrogenation enthalpy (delta_H), number of H2 molecules (nH2), and storage capacity (%wt H2). Additionally, the zip archive encompasses the source code (app.py) for an accompanying web application, available at https://qm9-lohc.streamlit.app/, and a Python script (query.py) designed for database querying. The interactive web app allows the user to select a range of dehydrogenation enthalpy, a range of hydrogen storage capacity, and the number of desired results. The app will then query the dataset and display molecules (in either 2D or 3D) along their hydrogen storage capacity and dehydrogenation enthalpy.
Technical Validation
The validation of our G4MP2 calculations is based in methodologies derived from prior research employing similar computational techniques49,50,51,59. Specifically, Narayanan et al.49 reported that the G4MP2 calculations yielded a mean absolute error (MAE) of 1.04 kcal/mol (4.35 kJ/mol) when compared with experimental gas-phase enthalpies of formation, showcasing their reliability and accuracy for the purposes of this study49. Additionally, Rogers et al. employed Gn methods to accurately calculate hydrogenation enthalpies of various small hydrocarbons, reporting MAEs between 3.5 and 5.0 kJ/mol65,66,67,68.
Due to the limited availability of experimental dehydrogenation enthalpies data for molecules within the QM9-LOHC dataset, the comparison was conducted on a set of 14 molecules (Table 1). The first nine molecules (Entries 1–9) in Table 1 are part of a benchmark set from our recent study19. The remaining entries (Entries 10–14) represent data obtained from the Pedley Dataset69 and the NIST workbook70, where we utilized the standard enthalpies of formation of the hydrogenated and dehydrogenated species to calculate the reaction enthalpies. This comparative analysis is shown in Table 1, showcasing the experimental dehydrogenation enthalpies alongside the corresponding values derived from QM9-LOHC via G4MP2 calculations. The findings reveal a close alignment between the computed values and experimental benchmarks, characterized by a root mean square deviation (RMSD) of 7.3 kJ/mol H2 and an MAE of 6.3 kJ/mol H2.
To better contextualize these results, we also performed G4 calculations for the same validation set (Table 1), obtaining a reduced RMSD of 4.6 kJ/mol H2 and an MAE of 2.9 kJ/mol H2. While the G4 method demonstrates improved accuracy on this small benchmark set, it comes at a significantly higher computational cost, making it less feasible for large-scale datasets such as QM9-LOHC. In contrast, the G4MP2 dataset offers a practical balance between computational efficiency and accuracy, with deviations that remain well within acceptable limits for quantum chemistry-based reaction studies. This highlights the suitability of the G4MP2 dataset as a reliable resource for hydrogenation and dehydrogenation reaction modeling in LOHC research.
We note that the observed discrepancies (up to 14.3 kJ/mol H2 for Aminobenzene) can be attributed to several factors. G4MP2, while reliable for many systems, is known to exhibit limitations for nitrogen-containing molecules, such as aniline derivatives71. Suntsova and Dorofeeva demonstrate that deviations for nitrogen species can exceed 10 kJ/mol, even with the higher-accuracy G4 method71. Furthermore, systematic underestimation in enthalpies of formation is observed for certain molecular classes, such as nitro compounds, with deviations ranging between 5–15 kJ/mol, as these classes were underrepresented in the original test sets used for method parameterization71,72. Finally, large deviations (>20 kJ/mol) may also reflect uncertainties or errors in the experimental reference data, as suggested by the isodesmic reaction validation method72.
Figure 3a presents the dehydrogenation enthalpies derived from QM9-G4MP2 for the QM9-LOHC dataset and F all the QM9-LOHC energy values. The left histogram includes 532 reactions from the original QM9-G4MP2 dataset that meet the Department of Energy’s 5.5% wt. H2 criterion. A majority of the reactions have dehydrogenation enthalpies above the optimal range for LOHC functionality, with most falling within 100–150 kJ/mol. In contrast, the right histogram, encapsulating a broader scope of 10,373 reactions in the QM9-LOHC dataset, identifies 3040 reactions with enthalpies between 40–70 kJ/mol, the optimal range for LOHC applications. Additionally, there are 423 reactions with dehydrogenation enthalpies below 40 kJ/mol and 5616 reactions with values between 70–120 kJ/mol (Fig. 3b). The QM9-LOHC dataset further reveals that the majority of molecules (8615), contain 9 heavy atoms, and 1114 comprise 8 heavy atoms (Fig. 3c). This distribution reflects a notable trend within the dataset: larger organic molecules, specifically those with 8 or 9 heavy atoms, are more likely to be liquid at room temperature—a key property for their function as LOHCs73.

(a) Histogram showing the distribution of dehydrogenation enthalpies (∆H) in QM9-LOHC dataset. (b) Pie chart showing the percentages and counts of reactions in the QM9-LOHC dataset that fall in the desired 40 – 70 kJ/mol H2 range as well as reactions with enthalpies less than 40 kJ/mol, 70–120 kJ/mol, and above 120 kJ/mol. (c) shows the distributions of heavy atoms in the QM9-LOHC dataset.
Figure 4 offers a nuanced view of the QM9-LOHC dataset, highlighting the interplay between dehydrogenation enthalpies and hydrogen storage capacities. Figure 4a shows the distribution of dehydrogenation enthalpies against hydrogen storage capacities, with the gold-shaded area in the figure indicating the optimal ∆H range (40–70 kJ/mol per H2) for conventional LOHCs. This visualization shows that that higher hydrogen storage capacities are generally associated with increased dehydrogenation enthalpies. Furthermore, it is apparent that fewer molecules reside within the gold-shaded region as storage capacity rises, illustrating a potential compromise between storage capacity and enthalpic efficiency.

(a) Scatter plot of hydrogen storage capacity and dehydrogenation enthalpy (∆H) across the dataset with a highlight on the preferred enthalpy range for conventional LOHCs (40–70 kJ/mol H2), (b) Pie chart showing the distribution of hydrogen storage capacity across the QM9-LOHC dataset (given in %wt. H2) and (c) shows four (A–D) random dehydrogenation reactions, dehydrogenation enthalpies, and hydrogen storage capacities. (shown in a. as the four red points).
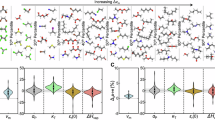
In Fig. 4b, the pie chart delineates the hydrogen storage capacity distribution within the dataset, revealing that a significant 62.5% of molecules have a capacity between 6 and 6.5%, with the next substantial group (19.9%) possessing a capacity between 7 and 8%. This data shows that the overwhelming majority of the dataset (95.5%) exceeds the DOE’s storage capacity threshold by at least 0.5%, affirming the dataset’s relevance in sourcing effective LOHCs. Representative reactions, denoted as Points A, B, C, and D in Fig. 4c, fall within the desired ∆H range and are depicted in Fig. 4c with varying hydrogen storage capacities. These points are characterized by diverse hydrogenation sites, including six-membered nitrogen-containing rings, carbonyl groups, azides, and five-membered rings with nitrogen and oxygen—each contributing to the molecular variety suitable for hydrogenation (Fig. 5).

Plots of selected features versus dehydrogenation enthalpies in the QM9-LOHC dataset (top left: number of oxygen atoms, top right: number of nitrogen atoms, bottom left: heavy atom count, bottom right: number of aromatic rings).
Code availability
The code and database of all G4MP2-calculated reaction enthalpies generated in this study are publicly available on GitHub: https://github.com/HydrogenStorage/QM9-LOHC. The data is also available on Zenodo: https://zenodo.org/records/10926772. A Jupyter notebook to query the QM9-LOHC database (requires pandas, rdkit, and numpy) is available on GitHub (https://github.com/HydrogenStorage/QM9-LOHC/blob/main/QM9-LOHC-Query.ipynb). We also present an interactive web app to navigate through the QM9-LOHC dataset and can be accessed through this link: https://qm9-lohc.streamlit.app. An overview of the G4MP2 method is available on the GitHub Repository.
References
Bockris, J. O. M. The Hydrogen Economy: Its History. International Journal of Hydrogen Energy 38(6), 2579–2588, https://doi.org/10.1016/j.ijhydene.2012.12.026 (2013).
Wei, D. et al. Toward a Hydrogen Economy: Development of Heterogeneous Catalysts for Chemical Hydrogen Storage and Release Reactions. ACS Energy Lett. 7(10), 3734–3752, https://doi.org/10.1021/acsenergylett.2c01850 (2022).
Preuster, P., Papp, C. & Wasserscheid, P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-Free Hydrogen Economy. Acc. Chem. Res. 50(1), 74–85, https://doi.org/10.1021/acs.accounts.6b00474 (2017).
Yap, J. & McLellan, B. A Historical Analysis of Hydrogen Economy Research, Development, and Expectations, 1972 to 2020. Environments 10(1), 11, https://doi.org/10.3390/environments10010011 (2023).
Wang, M., Wang, Z., Gong, X. & Guo, Z. The Intensification Technologies to Water Electrolysis for Hydrogen Production – A Review. Renewable and Sustainable Energy Reviews 29, 573–588, https://doi.org/10.1016/j.rser.2013.08.090 (2014).
Armor, J. N. The Multiple Roles for Catalysis in the Production of H2. Applied Catalysis A: General 176(2), 159–176, https://doi.org/10.1016/S0926-860X(98)00244-0 (1999).
Langmi, H. W., Engelbrecht, N., Modisha, P. M. & Bessarabov, D. Hydrogen Storage. In Electrochemical Power Sources: Fundamentals, Systems, and Applications; pp 455–486, https://doi.org/10.1016/B978-0-12-819424-9.00006-9 Elsevier, (2022).
Crabtree, R. H. Hydrogen Storage in Liquid Organic Heterocycles. Energy Environ. Sci. 1(1), 134, https://doi.org/10.1039/b805644g (2008).
Griffiths, S., Sovacool, B. K., Kim, J., Bazilian, M. & Uratani, J. M. Industrial Decarbonization via Hydrogen: A Critical and Systematic Review of Developments, Socio-Technical Systems and Policy Options. Energy Research & Social Science 80, 102208, https://doi.org/10.1016/j.erss.2021.102208 (2021).
Quarton, C. J. et al. The Curious Case of the Conflicting Roles of Hydrogen in Global Energy Scenarios. Sustainable Energy Fuels 4(1), 80–95, https://doi.org/10.1039/C9SE00833K (2020).
Allendorf, M. D. et al. Challenges to Developing Materials for the Transport and Storage of Hydrogen. Nat. Chem. 14(11), 1214–1223, https://doi.org/10.1038/s41557-022-01056-2 (2022).
Barthélémy, H. Hydrogen Storage – Industrial Prospectives. International Journal of Hydrogen Energy 37(22), 17364–17372, https://doi.org/10.1016/j.ijhydene.2012.04.121 (2012).
Langmi, H. W., Ren, J., North, B., Mathe, M. & Bessarabov, D. Hydrogen Storage in Metal-Organic Frameworks: A Review. Electrochimica Acta 128, 368–392, https://doi.org/10.1016/j.electacta.2013.10.190 (2014).
Aakko-Saksa, P. T., Cook, C., Kiviaho, J. & Repo, T. Liquid Organic Hydrogen Carriers for Transportation and Storing of Renewable Energy – Review and Discussion. Journal of Power Sources 396, 803–823, https://doi.org/10.1016/j.jpowsour.2018.04.011 (2018).
Chu, C., Wu, K., Luo, B., Cao, Q. & Zhang, H. Hydrogen Storage by Liquid Organic Hydrogen Carriers: Catalyst, Renewable Carrier, and Technology - A Review. Carbon Resources Conversion, S2588913323000248. https://doi.org/10.1016/j.crcon.2023.03.007 (2023).
Crabtree, R. H. Nitrogen-Containing Liquid Organic Hydrogen Carriers: Progress and Prospects. ACS Sustainable Chem. Eng. 5(6), 4491–4498, https://doi.org/10.1021/acssuschemeng.7b00983 (2017).
Niermann, M., Beckendorff, A., Kaltschmitt, M. & Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC) – Assessment Based on Chemical and Economic Properties. International Journal of Hydrogen Energy 44(13), 6631–6654, https://doi.org/10.1016/j.ijhydene.2019.01.199 (2019).
Rao, P. C. & Yoon, M. Potential Liquid-Organic Hydrogen Carrier (LOHC) Systems: A Review on Recent Progress. Energies 13(22), 6040, https://doi.org/10.3390/en13226040 (2020).
Harb, H. et al. Uncovering Novel Liquid Organic Hydrogen Carriers: A Systematic Exploration of Chemical Compound Space Using Cheminformatics and Quantum Chemical Methods. Digital Discovery. https://doi.org/10.1039/D3DD00123G (2023).
Teichmann, D., Arlt, W., Wasserscheid, P. & Freymann, R. A Future Energy Supply Based on Liquid Organic Hydrogen Carriers (LOHC). Energy Environ. Sci. 4(8), 2767–2773, https://doi.org/10.1039/C1EE01454D (2011).
Müller, K., Völkl, J. & Arlt, W. Thermodynamic Evaluation of Potential Organic Hydrogen Carriers. Energy Technology 1(1), 20–24, https://doi.org/10.1002/ente.201200045 (2013).
Cooper, A. C. Campbell, K. M. & Pez, G. P. An Integrated Hydrogen Storage and Delivery Approach Using Organic Liquid-Phase Carriers. (2006).
Modisha, P. M., Ouma, C. N. M., Garidzirai, R., Wasserscheid, P. & Bessarabov, D. The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers. Energy Fuels 33(4), 2778–2796, https://doi.org/10.1021/acs.energyfuels.9b00296 (2019).
Kariya, N., Fukuoka, A. & Ichikawa, M. Efficient Evolution of Hydrogen from Liquid Cycloalkanes over Pt-Containing Catalysts Supported on Active Carbons under “Wet–Dry Multiphase Conditions. Applied Catalysis A: General 233(1–2), 91–102, https://doi.org/10.1016/S0926-860X(02)00139-4 (2002).
Wild, J. et al. Liquid Organic Hydrogen Carriers (LOHC): An Auspicious Alternative to Conventional Hydrogen Storage Technologies. Energy Environ, 78 (2010).
Stark, K. et al. Melting Points of Potential Liquid Organic Hydrogen Carrier Systems Consisting of N -Alkylcarbazoles. J. Chem. Eng. Data 61(4), 1441–1448, https://doi.org/10.1021/acs.jced.5b00679 (2016).
Brodt, M. et al. The 2-Propanol Fuel Cell: A Review from the Perspective of a Hydrogen Energy Economy. Energy Technology 9(9), 2100164, https://doi.org/10.1002/ente.202100164 (2021).
Sievi, G. et al. Towards an Efficient Liquid Organic Hydrogen Carrier Fuel Cell Concept. Energy & Environmental Science 12(7), 2305–2314, https://doi.org/10.1039/C9EE01324E (2019).
Hauenstein, P., Seeberger, D., Wasserscheid, P. & Thiele, S. High Performance Direct Organic Fuel Cell Using the Acetone/Isopropanol Liquid Organic Hydrogen Carrier System. Electrochemistry Communications 118, 106786, https://doi.org/10.1016/j.elecom.2020.106786 (2020).
Yu, Y. et al. Reversible Hydrogen Uptake/Release over a Sodium Phenoxide–Cyclohexanolate Pair. Angewandte Chemie International Edition 58(10), 3102–3107, https://doi.org/10.1002/anie.201810945 (2019).
He, T., Cao, H. & Chen, P. The Roles of Alkali/Alkaline Earth Metals in the Materials Design and Development for Hydrogen Storage. Acc. Mater. Res. 2(9), 726–738, https://doi.org/10.1021/accountsmr.1c00048 (2021).
Kinetic Studies of Reversible Hydrogen Storage over Sodium Phenoxide-Cyclohexanolate Pair in Aqueous Solution. Journal of Energy Chemistry, 39, 244–248. https://doi.org/10.1016/j.jechem.2019.04.008 (2019).
Huang, B. & von Lilienfeld, O. A. Ab Initio Machine Learning in Chemical Compound Space. Chem. Rev. 121(16), 10001–10036, https://doi.org/10.1021/acs.chemrev.0c01303 (2021).
von Lilienfeld, O. A. Quantum Machine Learning in Chemical Compound Space. Angew Chem Int Ed 57(16), 4164–4169, https://doi.org/10.1002/anie.201709686 (2018).
Dral, P. O. Quantum Chemistry in the Age of Machine Learning. J. Phys. Chem. Lett. 11(6), 2336–2347, https://doi.org/10.1021/acs.jpclett.9b03664 (2020).
Lee, A. S. et al. E min: A First-Principles Thermochemical Descriptor for Predicting Molecular Synthesizability. J. Chem. Inf. Model. acs.jcim.3c01583. https://doi.org/10.1021/acs.jcim.3c01583 (2024).
Thakkar, A., Chadimová, V., Bjerrum, E. J., Engkvist, O. & Reymond, J.-L. Retrosynthetic Accessibility Score (RAscore) – Rapid Machine Learned Synthesizability Classification from AI Driven Retrosynthetic Planning. Chem. Sci. 12(9), 3339–3349, https://doi.org/10.1039/D0SC05401A (2021).
Voršilák, M., Kolář, M., Čmelo, I. & Svozil, D. SYBA: Bayesian Estimation of Synthetic Accessibility of Organic Compounds. J Cheminform 12(1), 35, https://doi.org/10.1186/s13321-020-00439-2 (2020).
Coley, C. W., Rogers, L., Green, W. H. & Jensen, K. F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 58(2), 252–261, https://doi.org/10.1021/acs.jcim.7b00622 (2018).
Abdalla, A. N. et al. Integration of Energy Storage System and Renewable Energy Sources Based on Artificial Intelligence: An Overview. Journal of Energy Storage 40, 102811, https://doi.org/10.1016/j.est.2021.102811 (2021).
Agarwal, G., Doan, H. A., Robertson, L. A., Zhang, L. & Assary, R. S. Discovery of Energy Storage Molecular Materials Using Quantum Chemistry-Guided Multiobjective Bayesian Optimization. Chem. Mater. 33(20), 8133–8144, https://doi.org/10.1021/acs.chemmater.1c02040 (2021).
Doan, H. A. et al. Quantum Chemistry-Informed Active Learning to Accelerate the Design and Discovery of Sustainable Energy Storage Materials. Chem. Mater. 32(15), 6338–6346, https://doi.org/10.1021/acs.chemmater.0c00768 (2020).
Luo, Z. et al. Survey of Artificial Intelligence Techniques Applied in Energy Storage Materials R&D. Frontiers in Energy Research, 8 (2020).
Patel, L., Shukla, T., Huang, X., Ussery, D. W. & Wang, S. Machine Learning Methods in Drug Discovery. Molecules 25(22), 5277, https://doi.org/10.3390/molecules25225277 (2020).
Paul, D. et al. Artificial Intelligence in Drug Discovery and Development. Drug Discovery Today 26(1), 80–93, https://doi.org/10.1016/j.drudis.2020.10.010 (2021).
Vamathevan, J. et al. Applications of Machine Learning in Drug Discovery and Development. Nat Rev Drug Discov 18(6), 463–477, https://doi.org/10.1038/s41573-019-0024-5 (2019).
Ruddigkeit, L., van Deursen, R., Blum, L. C. & Reymond, J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 52(11), 2864–2875, https://doi.org/10.1021/ci300415d (2012).
Ramakrishnan, R., Dral, P. O., Rupp, M. & Von Lilienfeld, O. A. Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci Data 1(1), 140022, https://doi.org/10.1038/sdata.2014.22 (2014).
Narayanan, B., Redfern, P. C., Assary, R. S. & Curtiss, L. A. Accurate Quantum Chemical Energies for 133 000 Organic Molecules. Chem. Sci. 10(31), 7449–7455, https://doi.org/10.1039/C9SC02834J (2019).
Curtiss, L. A., Redfern, P. C. & Raghavachari, K. Gaussian-4 Theory Using Reduced Order Perturbation Theory. The Journal of Chemical Physics 127(12), 124105, https://doi.org/10.1063/1.2770701 (2007).
Curtiss, L. A., Redfern, P. C. & Raghavachari, K. Gaussian-4 Theory. The Journal of Chemical Physics 126(8), 084108, https://doi.org/10.1063/1.2436888 (2007).
Curtiss, L. A., Redfern, P. C. & Raghavachari, K. G n Theory. WIREs Comput Mol Sci 1(5), 810–825, https://doi.org/10.1002/wcms.59 (2011).
Curtiss, L. A., Raghavachari, K., Redfern, P. C., Rassolov, V. & Pople, J. A. Gaussian-3 (G3) Theory for Molecules Containing First and Second-Row Atoms. The Journal of Chemical Physics 109(18), 7764–7776, https://doi.org/10.1063/1.477422 (1998).
Paragian, K., Li, B., Massino, M. & Rangarajan, S. A Computational Workflow to Discover Novel Liquid Organic Hydrogen Carriers and Their Dehydrogenation Routes. Mol. Syst. Des. Eng. 5(10), 1658–1670, https://doi.org/10.1039/D0ME00105H (2020).
Dandu, N. K. et al. Improving the Accuracy of Composite Methods: A G4MP2 Method with G4-like Accuracy and Implications for Machine Learning. J. Phys. Chem. A 126(27), 4528–4536, https://doi.org/10.1021/acs.jpca.2c01327 (2022).
Dandu, N. et al. Quantum-Chemically Informed Machine Learning: Prediction of Energies of Organic Molecules with 10 to 14 Non-Hydrogen Atoms. J. Phys. Chem. A 124(28), 5804–5811, https://doi.org/10.1021/acs.jpca.0c01777 (2020).
Sterling, T. & Irwin, J. J. ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 55(11), 2324–2337, https://doi.org/10.1021/acs.jcim.5b00559 (2015).
PubChem. PubChem. https://pubchem.ncbi.nlm.nih.gov/ (accessed 2023-10-09).
Dandu, N. K., Ward, L., Assary, R. S., Redfern, P. C. & Curtiss, L. A. Accurate Prediction of Adiabatic Ionization Potentials of Organic Molecules Using Quantum Chemistry Assisted Machine Learning. J. Phys. Chem. A 127(28), 5914–5920, https://doi.org/10.1021/acs.jpca.3c00823 (2023).
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. & Walsh, A. Machine Learning for Molecular and Materials Science. Nature 559(7715), 547–555, https://doi.org/10.1038/s41586-018-0337-2 (2018).
DOE Technical Targets for Onboard Hydrogen Storage for Light-Duty Vehicles. Energy.gov. https://www.energy.gov/eere/fuelcells/doe-technical-targets-onboard-hydrogen-storage-light-duty-vehicles (accessed 2023-03-27).
Hassan Harb. HydrogenStorage/QM9-LOHC: QM9-LOHC, https://doi.org/10.5281/ZENODO.10926772 (2024).
RDKit. https://www.rdkit.org/ (accessed 2023-01-05).
Frisch, M. J. et al. Gaussian 16, Gaussian, Inc., Wallingford CT (2016).
Amir Khairbek, A. Quantum Calculations to Estimate the Heat of Hydrogenation Theoretically. In Advanced Applications of Hydrogen and Engineering Systems in the Automotive Industry; Cocco, L., Aziz, M., Eds.; IntechOpen, https://doi.org/10.5772/intechopen.93955 (2021).
Li, Z., Rogers, D. W., McLafferty, F. J., Mandziuk, M. & Podosenin, A. V. Ab Initio Calculations of Enthalpies of Hydrogenation, Isomerization, and Formation of Cyclic C 6 Hydrocarbons. Benzene Isomers. J. Phys. Chem. A 103(3), 426–430, https://doi.org/10.1021/jp982997m (1999).
Rogers, D. W., McLafferty, F. J. & Podosenin, A. V. G2(MP2) and G2(MP2,SVP) Calculations of Enthalpies of Hydrogenation, Isomerization, and Formation of C 5 Hydrocarbons. 2. Substituted Cyclobutenes, Vinylcyclopropene, Spiropentane, and Methyltetrahedrane. J. Phys. Chem. A 102(7), 1209–1213, https://doi.org/10.1021/jp9731720 (1998).
Rogers, D. W., McLafferty, F. J. & Podosenin, A. V. Ab Initio Calculations of Enthalpies of Hydrogenation and Isomerization of Cyclic C 4 Hydrocarbons. J. Phys. Chem. 100(43), 17148–17151, https://doi.org/10.1021/jp961122+ (1996).
Pedley, J. B., Naylor, R. D. & Kirby, S. P. Thermochemical Data of Organic Compounds; Springer Netherlands: Dordrecht, https://doi.org/10.1007/978-94-009-4099-4 (1986).
Linstrom, P. NIST Chemistry WebBook, NIST Standard Reference Database 69, https://doi.org/10.18434/T4D303 (1997).
Suntsova, M. A. & Dorofeeva, O. V. Use of G4 Theory for the Assessment of Inaccuracies in Experimental Enthalpies of Formation of Aromatic Nitro Compounds. J. Chem. Eng. Data 61(1), 313–329, https://doi.org/10.1021/acs.jced.5b00558 (2016).
Dorofeeva, O. V., Kolesnikova, I. N., Marochkin, I. I. & Ryzhova, O. N. Assessment of Gaussian-4 Theory for the Computation of Enthalpies of Formation of Large Organic Molecules. Struct Chem 22(6), 1303–1314, https://doi.org/10.1007/s11224-011-9827-7 (2011).
Klein, D. R. Fourth edition. Organic Chemistry. (Wiley, Hoboken, NJ, 2021).
Konnova, M. E. et al. Thermochemical Properties and Dehydrogenation Thermodynamics of Indole Derivates. Ind. Eng. Chem. Res. 59(46), 20539–20550, https://doi.org/10.1021/acs.iecr.0c04069 (2020).
He, T., Pei, Q. & Chen, P. Liquid Organic Hydrogen Carriers. Journal of Energy Chemistry 24(5), 587–594, https://doi.org/10.1016/j.jechem.2015.08.007 (2015).
Clot, E., Eisenstein, O. & Crabtree, R. H. Computational Structure–Activity Relationships in H 2 Storage: How Placement of N Atoms Affects Release Temperatures in Organic Liquid Storage Materials. Chem. Commun. No. 22, 2231–2233. https://doi.org/10.1039/B705037B (2007).
Acknowledgements
This material is based upon work supported by Laboratory Directed Research and Development (LDRD) funding from Argonne National Laboratory, provided by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC02-06CH11357. We acknowledge the computing resources provided on “BEBOP” and “Improv”, two computing clusters operated by the Laboratory Computing Resource Center at Argonne National Laboratory (ANL). RSA would like to like acknowledge funding support from the Consortium for Computational Physics and Chemistry (CCPC), which is supported by the Bioenergy Technologies Office (BETO) of Energy Efficiency and Renewable Energy (EERE).
Author information
Authors and Affiliations
Contributions
H. Harb generated and parsed the computational dataset. H. Harb and S.N. Elliott performed the data analysis. The manuscript was written by H. Harb, S.N. Elliott, and R.S. Assary, with contributions and reviews from all other authors. The scope of the work was conceptualized by H. Harb, S.N. Elliott, L. Ward, and R.S. Assary. S.J. Klippenstein, L.A. Curtiss, and R.S. Assary secured the supporting funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Harb, H., Elliott, S.N., Ward, L. et al. Accurate Dehydrogenation Enthalpies Dataset for Liquid Organic Hydrogen Carriers. Sci Data 12, 171 (2025). https://doi.org/10.1038/s41597-025-04468-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04468-0
