
Abstract
Cyperus iria is an aggressive weed of rice throughout the world. Until now, the reference genome of C. iria has not been published. Here, we completed the chromosome-level genome assembly of C. iria based on Illumina, PacBio and Hi-C reads. The assembled genome size of C. iria was 479.08 Mb with a contig N50 of 7.02 Mb. 68 pseudochromosomes were produced using Hi-C scaffolding, accounting for 99.65% of the assembled genome. The number of predicted protein-coding genes is 47,395, of which 93.26% were annotated, and 37.69% repetitive sequences were identified. Our study provided a valuable genomic resource for the molecular biology research and the management of C. iria.
Similar content being viewed by others

Background & Summary
Agricultural weeds are a key constraint factor in agricultural production because they can compete with crops for light, nutrients, moisture and space, leading to decreased crop quality and yield1. Among all types of crop pests, weeds are known to cause the greatest reduction in crop yield2. As reported, the potential yield reduction caused by weeds could be up to 23%, 37%, 37%, 40%, 36% and 30% in wheat, soybeans, rice, maize, cotton and potatoes, respectively, with the average yield reduction of 34%, which is far more than the impacts of animal pests and pathogens3. With the increasing world population and decreasing available resources, weed management is a particularly important and challenging task.
From the evolutionary point, weeds is an excellent example of rapid adaptation to changing environments, due to their abundant genetic variation and plasticity, weeds can evolve more rapidly than crops4,5. Weeds have shown a remarkable capacity to quickly adapt to changing environmental factors, agricultural techniques, and weed-control strategies6. Understanding the evolution and adaptation of weeds is essential for effective weeds management. However, the genetic characterization of weed systems has received comparatively little attention7.
Genomic studies is an important approach to identify the origin of weed species and study their adaptive evolution, providing a basis for establishing effective weed management strategies8. However, despite the substantial impact of weeds on agricultural production, research on weeds have not received the necessary attention in both traditional molecular biology and genomic analyses7,9. To date, 2,847 plant species have been identified as weeds10. However, only approximately 26 species have been subjected to sequencing and de novo genome assembly9.
Cyperus iria (rice flatsedge), belonging to Cyperaceae family, is an annual sedge with a fibrous root system and C4 photosynthetic system11. It can reach 60 cm in height12. C. iria is native to tropical and subtropical countries13 with the ability to multiply rapidly and readily adapts to ecological niches14. The high seed production of C. iria (3000–5000 seeds per plant) combined with its short life cycle leads to a very high rate of reproduction15,16. C. iria is an aggressive weed throughout the world and has become a major agricultural weed in rice production systems in 22 countries16,17. It has been reported that a 64% reduction in rice yield is the result of C. iria infestation throughout the crop growth period18. Competition between C. iria and rice during the first 30 days can reduce the yield of rice by up to 12.9%, and infestation during the first 40 days can the yield of transplanted rice yield by up to 43.5%19. Chemical herbicides have become the preferred choice for C. iria control because it is highly efficient, less labour intensive, and cost effective. However, as a result of extensive herbicide use, C. iria has developed resistant populations to ALS (acetolactate synthetase) inhibitors, such as pyrazosulfuron-ethyl, halosulfuron-methyl and penoxsulam20. C. iria herbicidal resistance remains largely unknown at the molecular level. Therefore, understanding the function of C. iria genes and the mechanisms by which it has evolved to become invasive is critical to the management of this weed. In addition, as a traditional Chinese medicine, C. iria has been found to show promising pharmacological effects12,13,21,22. However, despite its medical importance, genomic and genetic information on C. iria is still very limited, severely hampering molecular and genetic research into this devastating weed.
Here, we generated a chromosome-level genome of the C. iria based on Illumina, PacBio and Hi-C technology. The assembled genome size of C. iria was 479.08 Mb (92.08% of the estimated genome size) with a contig N50 of 7.02 Mb. 99.65% of the assembled genome were anchored to 68 pseudochromosomes. The number of predicted protein-coding genes is 47,395, of which 93.26% were annotated, and 37.69% repetitive sequences were identified. The high-quality chromosome-level C. iria reference genome assembly, provides a strong basis for development of new strategies for the successful management of this aggressive weed in the future.
Methods
Plant material, library construction and sequencing
Seeds of the C. iria was collected from Huzhou, Zhejiang Province (30° 87′ N, 120° 10′ E) in 2021 and stored in 4 °C until used. Plants of C. iria were grown in small pots in the Experimental Greenhouse of Zhejiang University’s Zijingang Campus (30° 30′ N, 120° 08′ E) in Hangzhou, China, with 16 hours of light and temperatures of 25 °C at day and 20 °C at night. Fresh young leaves from the same one C. iria plant were collected and immediately frozen in liquid nitrogen. The cetyltrimethylammonium bromide (CTAB) method was employed to extract high quality, high molecular weight genomic DNA from young leaves23. The genomic DNA was used for the construction of an Illumina paired-end (PE) library with ∼450 bp insert sizes, and the libraries were constructed on an Illumina NovaSeq sequencing platform using Next-Generation Sequencing (NGS) following the standard procedure. For PacBio sequencing, the genomic DNA of C. iria was used for the construction of PacBio SMRTbell libraries according to the standard SMRTbell library preparation protocol, and the PacBio Sequel II platform was then utilized to sequence the libraries. The Hi-C library constructed from fresh young leaves of same one C. iria plant was then sequenced on an Illumina NovaSeq sequencing platform.
Total RNAs were extracted from four tissues (root, stem, leaf, and flower) of the same C. iria plant using TRIzol reagent. RNA from these four tissues was mixed equally for the construction of a PacBio Iso-Seq library. The cDNA was generated from mixed RNA using the SMARTer PCR cDNA Synthesis Kit (Clontech). The purified cDNA was then used to construct a Iso-Seq SMRTbell library using the SMRTbell Express Template Prep kit 1.0, which was subsequently sequenced on the PacBio Sequel II platform.
Genome survey
After removing low-quality reads, 10,000 high-quality data pairs were randomly selected and mapped to the NCBI nucleotide (NT) database, revealing the top five matched species. Jellyfish software (version 2.3.0)24 was used for k-mer analysis of all high-quality data. Based on the k-mer frequency analysis (k-mer = 19), genome characteristics including genome size, heterozygosity and repeat rate, were estimated by using GenomeScope25. The genome size was calculated as following Eq. (1):
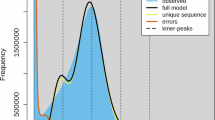
wherein G represents the genome size, N means the total number of reads, L means the average length of reads, K is the k-mer length, and D is the peak depth that is estimated from the k-mer distribution (pkdepth). Low frequency k-mers before the first valley were discarded to minimise the influence of sequencing errors. The k-mer analysis (k-mer = 19) analysis determined the genome size to be 520.28 Mb, with the heterozygosity rate and repetitive fraction were 0.08% and 47.23%, respectively (Table S1 and Fig. 1a). These findings suggest that the genome of C. iria belongs to the simple genome.

Genome size estimation and ploidy levels. (a) Estimation of genome size and heterozygosity based on k-mers (k = 19). (b) Ploidy inference based on Smudgeplot analysis.
Smudgeplot26 was used to estimate ploidy levels from corrected reads with a k-mer value of 19. The Smudgeplot performs gymnastics with heterozygous k-mer pairs extracted from k-mer count databases. It can disentangle genome structure by comparing the sum of coverages of k-mer pair (CovA + CovB) to their relative coverage (CovB/(CovA + CovB))26. Smudgeplot analysis indicates this genome assembly was a diploid (AB) with with a confidence of 0.91 (Fig. 1b).
Genome assembly
To obtain a contig-level genome, PacBio High Fidelity (HiFi) reads were assembled using Hifiasm27. Totally 18.51 Gb Pacbio HiFi reads were generated, resulting in a genome assembly of 479.08 Mb with a contig N50 of 7.02 Mb and a GC content of 35.64% (Tables 1, 2). Purge_dups (version 1.2.5)28 was used to remove redundant sequences in the polished assembly. After that, only 80 contigs (462.83 Mb) remained for further analysis (Table 3). BWA (version 0.7.12)29 was used to compare the corrected high-quality Illumina reads sequence with the assembled genome sequence. The results showed that the read sequence comparison rate reached 99.25%, and the average sequencing depth was 215×. The sequence depth of 20× or more accounted for 99.78% (Table 4).
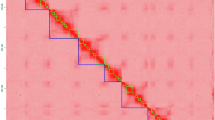
To obtain a chromosome-level genome assembly of the C. iria, HiC-Pro software (version 3.1.0)30 was used to align clean Hi-C reads to the draft genome sequences in comparison mode. All invalid read pairs were removed using HiC-Pro software except unique mapped paired-ends, which were retained for further analysis. In total, Hi-C sequencing produced 65.73 Gb clean reads (Table 5), and 99.15% of Hi-C reads mapped to assembled contigs, including 65.30% unique mapped read pairs (Table 5). PacBio sequencing assemblies were organized into chromosome-level scaffolds by integrating valid interaction pairs from the unique mapped read pairs (Table 5). The ensuing Hi-C reads were then used to correct misjoins, order, orient, and anchor in the draft genome assembly with the 3D de novo assembly (3D-DNA) (version 201008)31. We used the Hi-C data to attach the draft genome to the chromosome level, 68 pseudochromosomes were constructed, whereby 99.65% of the assembled sequences were anchored. The heatmap of Hi-C interaction revealed a higher intensity of interactions in diagonals is higher than that in nondiagonal positions in each group, indicating the chromosome-level genome assembly was complete and robust (Fig. 2a). Chromosome lengths varied from 3,585,826 bp (Chr54) to 10,521,918 bp (Chr66) (Table S2 and Fig. 2b). Subsequently, the assembled results was polished using Racon (version 1.4.20)32. Benchmark for Universal Single Copy Orthologues (BUSCO, version 5.4.3)33 with the plant dataset (embryophyte) was used to assess the genome completeness and continuity. BUSCO analysis showed that 95.11% (0.50% fragmented and 4.40% missing BUSCOs) of the BUSCO genes in the C. iria genome were successfully identified as complete BUSCO, which indicates the high completeness of the genome assembly (Table S3). Furthermore, the contiguity of the genome was evaluated by calculating LTR Assembly Index (LAI) using LTR_retriever (version 2.9.9)34 with default parameters. The LAI value of the genome assembly was 9.77.

Overview of the C. iria genome assembly. (a) Hi–C interaction heatmap for C. iria. (b) Chromosomal features of C. iria. The circle diagram from the outside to the inside represents: Pseudochromosome, Gene density, GC content, LTR/Gopia transposon density, LTR/Gypsy transposable factor density. The connection lines in the circle are synteny lines of paralogous sequences in the genome.
Genome annotation
To identify repeat sequences, a combination of the homology-based prediction and de novo-based prediction was performed. For de novo-based prediction, RECON (version 1.0.8)35, RepeatModeler (version 2.0.4)36, and RepeatScout (version 1.0.6)37 were used to identified repetitive elements. For homology-based prediction, RepeatMasker (version 4.1.4)38 was used to identify repetitive elements by comparing to consensus sequences in the Repbase library39. The analysis of homology-based comparisons and de novo-based methods revealed that 37.69% of the C. iria genome consists of repetitive sequences, with LTRs representing 8.95% of the total. Gypsy was identified as the predominant LTR type (5.85%), followed by Copia (2.69%) (Table 6).
Non-coding RNA (ncRNA) species, such as rRNA, tRNA, miRNA, and snRNA, were annotated with RNAmmer (version 1.2) (for rRNAs prediction)40, tRNAscan-SE (version 1.3.1) (for tRNAs prediction)41, and Perl program Rfam (version 1.0) (for other two non-coding RNAs prediction)42. Totally 2,355 ncRNA genes were also identified, which included 599 rRNA, 985 tRNA and 771 other ncRNA (Table 7).
To predict protein-coding genes, a combination of homology-based, de novo-based, and transcript-based prediction approaches was used. The de novo-based prediction was conducted with Augustus (version 3.3.2)43, GlimmerHMM (version 3.0.4)44, GeneMark (version 4.35)45, and GeneID (version 1.4) with default parameters. The homology-based prediction was performed using Exonerate (version 2.2.0)46 together with protein sequences from Carex littledalei, Arabidopsis thaliana and Oryza sativa. For transcript-based prediction, the high-quality full-length transcript data were compared and spliced using PASA (version 2.5.2)47 to obtain the corresponding gene prediction results. Finally, EvidenceModeler (version r2012-06-25)48 was used to integrate homology-based, de novo-based, and transcript-based prediction results. The prediction results revealed that C. iria genome contained 47,395 protein-coding genes, and the average gene length is 2,762.5 bp (Table 8). The BUSCO analysis evaluated the gene set completeness, revealing that 96.28% (1,554 genes) of the BUSCO genes were present in the C. iria gene set. This finding confirms the high quality of gene prediction. BLASTP (version 2.0.14.152) (E-value < 1 × 10−5) searches against the NR and SwissProt databases were performed for the final protein-coding, functionally annotated genes. Functional domains were then obtained by searching publicly available databases using InterProScan (version 5.61–93.0)49. GO annotation of protein-coding genes was performed using interproscan. The KEGG pathway annotation of protein-coding genes was mainly performed by KEGG Automatic Annotation Server (KAAS, version 2.1)50. Approximately 93.26% of the protein-coding genes were functionally annotated (Table 9). To further validate the accuracy of our gene annotations, we compared the gene sequences with transcript sequences using geneBody_coverage (version 5.0.1)51. This analysis assessed the sequence coverage across the entire gene length, from the 5′ to the 3′ end, to determine if the sequencing reads were evenly distributed. Figure S1 illustrates the alignment results, demonstrating that the sequencing reads were uniformly distributed without bias towards the 5′ or 3′ ends. The mapping rate of transcript sequences to the gene sequences was 93%, further supporting the accuracy of the gene annotations.
Data Records
Raw Illumina, PacBio HiFi and Hi-C of C. iria genome sequencing data were deposited in the NCBI BioProject database under project accession number PRJNA115799452, with accession numbers SRR3058810953 for Illumina sequencing data, SRR3058810854 for Pacbio sequencing data, SRR3058810755 for Hi-C sequencing data, SRR3058810656 for Iso-Seq data. The genome assembly has been deposited at GenBank under the accession JBHOFI00000000057. The annotation of the C. iria genome has been submitted to the online open-access repository Figshare58 database.
Technical Validation
BWA (version 0.7.12)29 was used to compare the corrected high-quality Illumina reads sequence with the assembled genome sequence. The read sequence comparison rate reached 99.25%, and the average sequencing depth was 215×. The sequence depth of 20× or more accounted for 99.78% (Table 4). Benchmark for Universal Single Copy Orthologues (BUSCO, version 5.4.3)33 with the plant dataset (embryophyte) was used to assess the genome completeness and continuity. BUSCO analysis showed that 95.11% (0.50% fragmented and 4.40% missing BUSCOs) of the BUSCO genes in the C. iria genome were successfully identified as complete BUSCO, which indicates the high completeness of the genome assembly (Table S3). Then, the assembly continuity was determined by analyzing the LTR Assembly Index (LAI), the LAI score was 9.77. The heatmap of Hi-C interaction revealed a higher intensity of interactions in diagonals is higher than that in nondiagonal positions in each group, indicating the chromosome-level genome assembly was complete and robust (Fig. 2a). The BUSCO analysis also used to evaluate the gene set completeness, revealing that 96.28% (1,554 genes) of the BUSCO genes were present in the C. iria gene set. This finding confirms the high quality of gene prediction.
Code availability
The pipeline and software utilized in this study were employed for data analysis following manual instructions and protocols. Details on the software version and parameters are outlined in the Methods section. In cases where specific parameters are not specified, default settings were applied.
References
Zhu, J. et al. Weed research status, challenges, and opportunities in China. Crop Prot. 134, 104449 (2020).
Jabran, K., Mahajan, G., Sardana, V. & Chauhan, B. S. Allelopathy for weed control in agricultural systems. Crop Prot. 72, 57–65 (2015).
Oerke, E.-C. Crop losses to pests. J. Agric. Sci. 144, 31–43 (2006).
Vercellino, R. B., Hernández, F., Pandolfo, C., Ureta, S. & Presotto, A. Agricultural weeds: the contribution of domesticated species to the origin and evolution of feral weeds. Pest Manag. Sci. 79, 922–934 (2023).
Vigueira, C. C., Olsen, K. M. & Caicedo, A. L. The red queen in the corn: agricultural weeds as models of rapid adaptive evolution. Heredity 110, 303–311 (2013).
Baucom, R. S. Evolutionary and ecological insights from herbicide-resistant weeds: what have we learned about plant adaptation, and what is left to uncover? New Phytol. 223, 68–82 (2019).
Guo, L. et al. Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nat. Commun. 8, 1031 (2017).
Wang, L. et al. Genomic insights into the origin, adaptive evolution, and herbicide resistance of Leptochloa chinensis, a devastating tetraploid weedy grass in rice fields. Mol. Plant 15, 1045–1058 (2022).
Huang, Y. et al. Weed genomics: yielding insights into the genetics of weedy traits for crop improvement. Abiotech 4, 20–30 (2023).
Weed Science Society of America. https://wssa.net/ (2024).
Riar, D. S. et al. Acetolactate Synthase-Inhibiting, Herbicide-Resistant Rice Flatsedge (Cyperus iria): Cross-Resistance and Molecular Mechanism of Resistance. Weed Sci. 63, 748–757 (2015).
Saeed, M. et al. Cyperus iria aqueous-ethanol extract ameliorated hyperglycemia, oxidative stress, and regulated inflammatory cytokines in streptozotocin-induced diabetic rats. Environ. Sci. Pollut. Res. 29, 4769–4784 (2022).
Roy, R., Roy, J., Liya, I. J., Basher, M. A. & Miah, M. Y. Antipyretic and antinociceptive effects of methanolic extract of C. iria L. tuber. J. Ethnopharmacol. 318, 116860 (2024).
Yang, L., Niu, J. & Tang, W. The complete chloroplast genome of pioneering plant Cyperus iria L. (Cyperaceae) in ecological restoration. Mitochondrial DNA Part B-Resour. 6, 1335–1336 (2021).
Awan, T. H., Ali, H. H. & Chauhan, B. S. Cyperus iria Weed Growth, Survival, and Fecundity in Response to Varying Weed Emergence Times and Densities in Dry-Seeded Rice Systems. Agron.-Basel 12, 1006 (2022).
Chauhan, B. S. & Johnson, D. E. Ecological studies on Cyperus difformis, Cyperus iria and Fimbristylis miliacea: three troublesome annual sedge weeds of rice. Ann. Appl. Biol. 155, 103–112 (2009).
Awan, T. H., Sta Cruz, P. C. & Chauhan, B. S. Growth analysis and biomass partitioning of Cyperus iria in response to rice planting density and nitrogen rate. Crop Prot. 74, 92–102 (2015).
Ismail, B. S. & Siddique, M. A. B. The Inhibitory Effect of Grasshopper’s Cyperus (Cyperus iria L.) on the Seedling Growth of Five Malaysian Rice Varieties. Trop. Life Sci. Res. 22, 81–89 (2011).
Dhammu, H. & Sandhu, K. S. Critical period of Cyperus iria L. competition in transplanted rice. Proc. 13th Aust. Weed Conf. Northam West. Aust. 79–82 (2013).
The International Survey of Herbicide Resistant Weeds. https://www.weedscience.org/Home.aspx (2024).
de Vera, P. J. D., Tayone, J. C. & De las Llagas, M. C. S. Cyperus iria linn. Roots ethanol extract: its phytochemicals, cytotoxicity, and anti-inflammatory activity. J. Taibah Univ. Sci. 16, 854–862 (2022).
Vicencio, E. J. M. & Buot, I. E. Weed research in asean countries: an annotated bibliography. J. Nat. Stud. 16, 24–53 (2017).
Healey, A., Furtado, A., Cooper, T. & Henry, R. J. Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 10, 1–8 (2014).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170‐+ (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinforma. Oxf. Engl. 36, 2896–2898 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 25, 1754–1760 (2009).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Vaser, R., Sovic, I., Nagarajan, N. & Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinforma. Oxf. Engl. 31, 3210–3212 (2015).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Bao, Z. R. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457 (2020).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, I351–I358 (2005).
Tempel, S. Using and understanding RepeatMasker. Methods Mol. Biol. Clifton NJ 859, 29–51 (2012).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffiths-Jones, S. Annotating non-coding RNAs with Rfam. Curr. Protoc. Bioinforma. Chapter 12, (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM:: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Besemer, J., Lomsadze, A. & Borodovsky, M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618 (2001).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
NCBI BioProject. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1157994 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30588109 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30588108 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30588107 (2024).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR30588106 (2024).
NCBI GenBank. https://identifiers.org/ncbi/insdc:JBHOFI000000000 (2024).
Yang, S. Genome assembly and annotation of Cyperus iria. Figshare https://doi.org/10.6084/m9.figshare.27014713 (2024).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32072433) and the Innovation and Development Special Fund for the West Hangzhou Science and Technology Innovation Corridor.
Author information
Authors and Affiliations
Contributions
W.G. and J.Z. devised the study and conceived the project. Q.C. and Z.W. collected the samples. S.Y., Q.C., Z.W., A.W., S.S., Z.L. participated in date analyses. S.Y. wrote the manuscript. S.L., W.G. and J.Z. approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, S., Cao, Q., Wen, Z. et al. Chromosome-level genome assembly of Cyperus iria, an aggressive weed of rice. Sci Data 12, 120 (2025). https://doi.org/10.1038/s41597-025-04470-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04470-6
