
Abstract
The Multidisciplinary Observatory for Study of the Arctic Climate (MOSAiC) expedition consisted of a year-long drifting survey of the Central Arctic Ocean. The ecosystems component of MOSAiC included the sampling of molecular data, with metagenomes collected from a diverse range of environments. The generation of metagenome-assembled-genomes (MAGs) from metagenomes are a starting point for genome-resolved analyses. This dataset presents a catalogue of MAGs recovered from a set of 73 samples from MOSAiC, including 2407 prokaryotic and 56 eukaryotic MAGs, as well as annotations of a near complete eukaryotic MAG using the Joint Genome Institute (JGI) annotation pipeline. The metagenomic samples are from the surface ocean, chlorophyll maximum, mesopelagic and bathypelagic, within leads and under-ice ocean, as well as melt ponds, ice ridges, and first- and second-year sea ice. This set of MAGs can be used to benchmark microbial biodiversity in the Central Arctic Ocean, compare individual strains across space and time, and to study changes in Arctic microbial communities from the winter to summer, at a genomic level.
Similar content being viewed by others

Background & Summary
The Central Arctic Ocean is one of the most understudied biomes on Earth and it is home to a significant amount of microbial diversity relative to its area. Furthermore, the Arctic Ocean is warming at a higher rate than lower latitudes, which has detrimental effects on the diversity of Arctic ocean biomes1. Microbes that underpin these biomes play an important role as primary producers and therefore as the base of the marine food web and central for biogeochemical cycles. They are also a source of novel genes, many of which have been of interest to the biotechnology industry2. It is therefore important to understand the effect of changing environmental conditions on these organisms including the evolution of their genes and genomes. However, due to the inaccessibility of the Central Arctic Ocean, there is a large gap in our understanding of microbial communities which thrive in this habitat. The MOSAiC expedition provided invaluable opportunities to address this knowledge gap and is the first year-long survey of the Central Arctic Ocean, facilitating sampling throughout the entire Arctic winter.
The MOSAiC expedition (2019–2020) was a Lagrangian drift survey, based around the icebreaker RV Polarstern3, which moved from north of the Laptev Sea northwards during leg 1 (19th September to 15th December 2019), before drifting southwards toward the Fram Strait (legs 2 to 4, 15th December 2019 to 12th August 2020) and finally returning to the Central Arctic Ocean (leg 5, 12th August to 12th October 2020) at the end of the expedition. During each leg, metagenomes were collected from both sea ice and pelagic waters, with a minority of samples collected from sediment traps under sea ice. Samples were collected either as part of a core time series, from intense observation periods of opportunistic sampling, or as part of the HAVOC project (Ridges – safe HAVens for ice-associated flora and fauna in a seasonally ice-covered Arctic Ocean), collecting samples from ice-ridges, under-ice water, and from sediment traps beneath sea-ice ridges and level ice4.
Sea-ice ridges are characteristic features covering 25 to 45% of the Arctic sea ice area5. Ridges are formed by pressure from drifting ice. When ice floes are forced together, they break up and are pushed up and down to form a sail above and a keel below the surface water level. The keel consists of ice blocks separated by voids, described as macroporosity, making up ~15–30% of the volume6,7. The voids may be empty (in the sail) or filled with liquid or frozen seawater or meltwater (in the keel). While the bottom of level sea ice is known as an important habitat for Arctic marine biodiversity and activity, much less is known of the life within ridge keel voids which constitute unique habitats and biological hotspots in the Arctic Ocean8,9,10. As ridges are logistically harder to navigate and take samples from, they are relatively understudied compared to level ice. The aim of the HAVOC project was to better understand how ice ridges act as a refuge for microbial biodiversity and activity, and how food web and biogeochemical processes at the ice-ocean interface and the underlying water column differ between ridges and level sea ice4.
This study consists of metagenome-assembled-genomes (MAGs) assembled from a pilot sequencing project of 15 metagenomes collected during Leg 2 (15th December to 3rd March) of the voyage in the Arctic winter as part of the core time series, and 58 metagenomes associated with the HAVOC project. Advances in metagenomic sequencing, assembly and binning, have generated a wealth of MAG datasets, even for Arctic environments such as in11. However, challenges associated with the assembly and binning of eukaryotes have meant that these generally focus on prokaryotic MAGs. Two exceptions are Duncan et al. and Delmont et al.12,13, which generated 21 and 25 eukaryotic medium and high quality MAGs from Arctic metagenomes respectively (i.e. above 50% completeness). The data presented here includes both prokaryotic and eukaryotic MAGs, using coassembly to improve the coverage of eukaryotes. These MAGs can be used to gain insights into microbial diversity and the metabolic potential of microbiomes during the Arctic winter, and the role of ice ridges in maintaining the microbial biodiversity of the Arctic Ocean.
Methods
Sampling
Of the 73 samples presented here, 15 were collected as part of a year-round time-series, during leg 2 of the expedition (between 13 January 2019 and 7 February 2020) as described in Winder et al.14, and sequenced as part of a pilot sequencing project (hereon called pilot samples). Of these samples, 8 were from pelagic layers and the remaining 7 from sea ice. The pelagic pilot samples were collected using a CTD rosette, on three different days. Sequenced pilot samples from a sampling event on February 6th 2020 consist of 2 co-located samples (i.e. replicates, from the same CTD cast and sampled at the same depth, but from different Niskin bottles) taken from a depth of 20 m, and one sample from a depth of 202 m. One sample was collected on February 7th 2020, at a depth of 4082 m. Further, 2 replicates from a depth of 50 m sampled on January 16th 2020 were sequenced.
Additionally, for each of the two biological replicates, a third technical replicate was generated by pooling remaining material from the two replicate samples. These data are summarised in Supplementary Table 1, with the two samples generated through pooling identified with a sample identifier suffix ‘pool’ (column E).
The remaining 7 pilot samples from sea ice were taken from the first-year and second-year MOSAiC ice-coring sites on the floe, as described in Nicolaus et al.15 and Fong et al.16. First and second-year sea-ice chemical and physical properties are available in Oggier et al.17,18, Lei et al.19, and properties for snow in Macfarlane et al.20. Generally, 2–4 cores were collected on a weekly basis, cut in 10 cm sections, except the bottom where two 5 cm thick section were cut, and pooled per section to allow for enough biomass in the DNA samples. For the pilot study, samples per depth interval always represent pools of three individual cores collected after each other in the same location (adjacent within 40 cm). Of these, five samples were collected from different 5–10 cm thick sections of cores from the same coring site on February 3rd 2020 from first-year ice; three from the upper part (20–50 cm from the top), one from the middle section (70–80 cm from the top), and one from the bottom-most section (122 to 127 cm from the top) of the sea ice, i.e. at the sea-ice interface. The remaining two samples were second-year ice, also from the bottom most 5 cm section, i.e. from the sea–ice interface, 1.23–1.28 m and 1.43–1.48 m from the top, collected on January 13th and 27th 2020.
The 58 metagenomes from the HAVOC project were collected during legs 2, 3 and 4 (collection dates between 22nd January 2020 and 26th July 2020), either: from sediment traps, directly below an ice ridge (7 samples) or level ice (10 samples) at depths of 5, 15 and 50 m, in the water column at 20 m (2 samples), from under ice water below an ice ridge (2 samples) and level ice (3 samples), from seawater taken from voids in the ice ridge (10 samples), or from a 10 cm ice core section at different depths of the ice ridge, as either ridge bottom ice (3 samples), top of void ice (5 samples), bottom of void ice (6 samples), refrozen void ice (4 samples) and ice samples at irregular depths (6 samples). Samples from the same location, depth, and time (Supplementary Table 1) are considered replicates, with samples taken from a total of 24 distinct locations and depths. The number of pooled core sections for each sample, and section thickness, are recorded in Supplementary Table 1. Figure 1 summarises the locations of the samples.

Map showing the locations of the samples. Panel A shows the overall course of the drift, and locations of the samples. Panel B shows more detailed locations, zoomed within the boxes marked in panel A. Samples are from leg 2 (15th Dec. 2019–3rd March 2020), leg 3 (3rd March–6th June 2020), and leg 4 (6th June–12th August 2020), with the drift route generally moving southward from the Central Arctic Ocean. Often, multiple (replicate) samples are co-located, either from the same CTD cast, or as different layers within a single ice core. In panel B the number of co-located samples is represented by the size of the marker.
For both the HAVOC and pilot sea ice samples, ice cores were sectioned in the field, transferred to sterile plastic bags, and brought back to the RV Polarstern. On board, 50 ml 0.22 µm filtered sea water was added per cm sea ice, and the sea ice samples melted within 24–36 hours in the dark at around 17–22°C. The use of filtered seawater was made necessary due to logistical constraints during the drift; this is a possible source of contamination for the sea ice samples. For both sea ice and pelagic samples, water was filtered through a Sterivex 0.22 µm filter or when volumes were <500 mL (HAVOC sediment trap samples and three HAVOC ice samples) onto 0.22 µm Durapore filters. The filters were immediately flash frozen in liquid nitrogen and stored at −80 °C on board the RV Polarstern, and subsequently shipped to either the Alfred Wegener Institute (pilot samples), or the University of Bergen (HAVOC samples), at a temperature of −80 °C.
DNA extraction, purification, and sequencing
Following shipping, DNA from Sterivex filters was extracted using the Qiagen PowerWater DNA kit, following the QIAGEN DNeasy Power Water SOP v1 for the ice and water samples, and the QIAGEN Dneasy Power Soil SOP v1 (QIAGEN N.V., Hilden, Germany) used for the DNA extraction from Durapore filters.
Plates were shipped to the Joint Genome Institute (JGI, CA, USA) under dry ice, and sequenced using 151 base pair paired-end reads with an Illumina NovaSeq S4 device. The library type used was in all cases either the Illumina low or regular concentration protocol, with between 0 and 15 rounds of PCR applied to samples, though in all but 5 cases, this was restricted to only either 0 or 5 rounds.
Supplementary Table 3 outlines the library preparation steps and sequencing protocols used for each of the samples.
Genome assembly and binning
Samples were first assembled individually using the JGI MAP pipeline21, with prokaryotic bins recovered on a per-sample basis. In brief, samples were filtered for quality with BBDuk, error corrected using BBCMS, assembled using SPAdes22, and reads mapped back to contigs using BBMap (38.86)23. Binning was performed using MetaBAT 224 (v2.1.15) and assessed for quality using CheckM25 (v1.1.3). Software and pipeline versions used are listed in Supplementary Table 3.
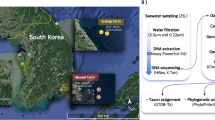
To extract further eukaryotic bins, we used a coassembly method. We used a custom filtering pipeline to identify the eukaryotic fraction of reads from each sample, before pooling these reads for coassembly and binning. To extract the eukaryotic fraction of reads, we used MMSeqs226 (version 0188988235c6f1a8e90f327827c73f981db8a19a) with the default parameters (length cutoff of 500 base pairs) to taxonomically identify contigs that had already been assembled using a per-sample assembly method, using a combination of MMETSP27 and NCBI NR28 as a reference database. Contigs identified at the domain level as anything other than Bacteria, Archaea, or viruses were retained, leaving a list of putatively eukaryotic contigs. Contigs identified as belonging to already existing prokaryotic bins were removed from this list. Next, the quality-filtered reads were mapped to this subset of contigs with BBMap (v.3.17). These reads were pooled, depending on whether they were from the HAVOC or pilot dataset, and assembled with metahipmer29 (version 2.1.0.1.380-gf770aca-dirty-master) for the HAVOC samples, or SPAdes (v3.14.0) with the metaspades.py–only-assembler option for the pilot samples. Samples with no pre-existing metagenome bins from their single assembly were excluded. Finally, the pooled reads from each dataset were mapped to their respective coassemblies, and then the new contigs were binned using MetaBAT 2 (version 2:v2.15-30-g4ec2ab8), and checked for quality with EukCC30 (2.1.1, database version 1.1). Bins of over 90% completeness and less than 5% contamination, and with at least 18 tRNA genes, and with 23S, 16S and 5S genes, were designated as high-quality MAGs, those with above 50% completeness and less than 10% contamination were designated medium-quality, and, for the eukaryotic MAGs, those above 30% completeness and less than 10% contamination were retained and designated as low quality, as per Alexander et al.31. Figure 2 shows a schematic diagram of the bioinformatics pipeline, and an overview of MAG completeness and contamination is provided in Fig. 3.

A summary of the IMG metagenome annotation pipeline, and the coassembly pipeline used for the two sample sets; either the pilot samples or the HAVOC samples. Coloured boxes indicate intermediate folders or files, either one per sample in the case of the stacked boxes, or one for each sample set, in the case of the coassemblies. Arrows indicate which files are inputs and outputs for other processes.

Completeness and contamination of the 2463 MAGs recovered across the 73 samples; 2407 prokaryotic and 56 eukaryotic MAGs. In each panel, a vertical line separates the eukaryotic and prokaryotic MAGs. The number of MAGs per taxon is shown between the two boxplots.
Functional and taxonomic annotation
MAGs recovered from single-sample assemblies were annotated using the IMG/M annotation pipeline (versions ranging between 5.0.23 and 5.1.11), using Genemark (v1.05) and Prodigal (V2.6.3)32,33 for gene calling, and HMMer34 (3.1b2) to combine analyses from the COG, PFam35 (v.34), TIGRFAM36 (v.15.0), Cath-Funfam37 (v.4.1.0), SuperFamily38 (v.1.75), and SMART39 (01_06_2016) databases. CRISPRs, and tRNAs were identified with CRT40 (1.8.2) and tRNAscan-SE41 (2.0.4) respectively. Prokaryotic MAGs were taxonomically placed with GTDB-tk (v.2.4.0, database release 220)42. GO terms were included based on the PFam2GO mapping provided by Interpro43,44.
To identify genes within the coassembled eukaryotic MAGs within the coassemblies, we used MetaEuk45 (version f32e8dfc6b994025627326d0f461f3e9903e997e) with the–easy-annotate option, using a custom database of the combined Phycocosm46, MMETSP, and UniRef47 databases, with UniRef clustered at a 50% identity level. These were combined with genes identified through Genemark-ES (version 4.71_lic, gmes_pepal.pl–ES), with genes from MetaEuk given priority and retained if overlapping with genes from Genemark-ES. PFam (v.35.0), PANTHER (v.17.0), SMART (v.9.0), NCBIfam (12.0) and SuperFamily (v.1.75) domains were then annotated using InterProScan48 (version 5.63–95.0).
Eukaryotic MAGs were placed on a phylogenetic tree (Fig. 4), using a set of 100 concatenated BUSCO49 genes (BUSCO v5.1.1 odb_eukaryota_10 gene set, aligned using MUSCLE50 v3.8.1551), alongside a set of 140 eukaryotic reference genomes from Phycocosm and NCBI RefSeq28. A maximum-likelihood tree was generated using FastTree (2.1.11).

Phylogenetic trees showing MAGs from singly assembled samples (left) and MAGs from coassemblies (right). Reference genomes, common to both trees, have their leaves aligned and are labelled at the centre of the tree, with collapsed clades represented in the tree by a wedge. Some leaves of reference genomes are unlabelled for legibility, where they share their genus with their closest relative in the tree. MAGs labelled are shown on the tips of each tree. For each MAG, the colour of the background indicates the type of the sample (or indeterminate type, in the case of the coassemblies). The coassembled MAG havoc.90, marked with an asterisk *, had good completeness and contamination scores and was subsequently renamed Bacilliariophyceae sp. MOSAICH1_1.
Case-Study of a high-quality MAG
From our eukaryotic coassembly, we flagged bin havoc.90 from the HAVOC dataset as having good contamination and completeness scores, as measured by BUSCO (v5.1.1, odb_eukaryota_10 gene set), with completeness and contamination of 73% and 0.4% respectively. This MAG, Bacilliariophyceae sp. MOSAICH1_1, was annotated using the Phycocosm pipeline; in brief, scaffolds were masked for repeats with RepeatMasker, and genes were called using a combination of Genemark-ES (version 4), GeneWise51 (version 1), and fgenesh52 (fgenesh1_pg), with the best fitting model for each locus picked to form a set of filtered gene models. Genes were functionally annotated for signal peptides, transmembrane domains, and assigned functional descriptions including assignment of PFam, GO, KOG and KEGG terms, and genes were formed into gene clusters using the MCL algorithm53.
This genome had a length of 32.07 Mbp, contained in 2963 scaffolds, with 13169 genes called among the set of filtered gene models. The most closely related isolate genome in Phycocosm was the diatom Pseudo-nitzschia multiseries CLN-47, with an average nucleotide identity of 76% (estimated with fastANI v1.33).
Data Records
All MAGs are available through Figshare54, at NCBI BioProject PRJNA1160706 (Data Citation 2)55, and replicated in the GOLD database56 (https://gold.jgi.doe.gov), Study ID 505419. Annotations of Bacilliariophyceae sp. MOSAICH1_1 are also available through the Phycocosm web portal (https://phycocosm.jgi.doe.gov/Mosaich1_1/Mosaich1_1.home.html), and via Figshare.
Individual read files for samples are stored in the NCBI SRA, with BioSample, BioProject, and SRP accessions listed in Supplementary Table 1, and as citations57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129.
Technical Validation
Prokaryotic MAG completeness and contamination was estimated using CheckM25, and only those MAGs within the MIMAG standards of 50% completeness and below 10% contamination were retained. Eukaryotic MAG completeness and contamination was estimated using EukCC – those above 30% completion and less than 10% contamination were retained, with those under 50% completion designated as low-quality MAGs.
Code availability
The custom pipelines used for eukaryotic MAG binning and annotation are available at https://github.com/willboulton/mosaic-pilot-havoc-mags.
References
Ardyna, M. & Arrigo, K. R. Phytoplankton dynamics in a changing Arctic Ocean. Nat. Clim. Chang. 10, 892–903 (2020).
Dalmaso, G. Z. L., Ferreira, D. & Vermelho, A. B. Marine Extremophiles: A Source of Hydrolases for Biotechnological Applications. Mar Drugs 13, 1925–1965 (2015).
Knust, R. Polar Research and Supply Vessel POLARSTERN Operated by the Alfred-Wegener-Institute. Journal of large-scale research facilities JLSRF 3, A119–A119 (2017).
Granskog, M. & Müller, O. A peek beneath the surface of Arctic sea ice. EU RES 37, 38–39 (2024).
Mårtensson, S., Meier, H. E. M., Pemberton, P. & Haapala, J. Ridged sea ice characteristics in the Arctic from a coupled multicategory sea ice model. Journal of Geophysical Research: Oceans 117 (2012).
Timco, G. W. & Burden, R. P. An analysis of the shapes of sea ice ridges. Cold Regions Science and Technology 25, 65–77 (1997).
Strub-Klein, L. & Sudom, D. A comprehensive analysis of the morphology of first-year sea ice ridges. Cold Regions Science and Technology 82, 94–109 (2012).
Gradinger, R., Bluhm, B. & Iken, K. Arctic sea-ice ridges—Safe heavens for sea-ice fauna during periods of extreme ice melt? Deep Sea Research Part II: Topical Studies in Oceanography 57, 86–95 (2010).
Syvertsen, E. E. Ice algae in the Barents Sea: types of assemblages, origin, fate and role in the ice-edge phytoplankton bloom. Polar Research 10, 277–288 (1991).
Fernández-Méndez, M. et al. Algal Hot Spots in a Changing Arctic Ocean: Sea-Ice Ridges and the Snow-Ice Interface. Frontiers in Marine Science 5 (2018).
Royo-Llonch, M. et al. Compendium of 530 metagenome-assembled bacterial and archaeal genomes from the polar Arctic Ocean. Nat Microbiol 6, 1561–1574 (2021).
Duncan, A. et al. Metagenome-assembled genomes of phytoplankton microbiomes from the Arctic and Atlantic Oceans. Microbiome 10, 67 (2022).
Delmont, T. O. et al. Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean. Cell Genomics 2, 100123 (2022).
Winder, J. C. et al. Genetic and Structural Diversity of Prokaryotic Ice-Binding Proteins from the Central Arctic Ocean. Genes 14, 363 (2023).
Nicolaus, M. et al. Overview of the MOSAiC expedition: Snow and sea ice. Elementa: Science of the Anthropocene 10, 000046 (2022).
Fong, A. A. et al. Overview of the MOSAiC expedition: Ecosystem. Elementa: Science of the Anthropocene 12, 00135 (2024).
Oggier, M. et al. First-year sea-ice salinity, temperature, density, oxygen and hydrogen isotope composition from the main coring site (MCS-FYI) during MOSAiC legs 1 to 4 in 2019/2020. PANGAEA https://doi.org/10.1594/PANGAEA.956732 (2023).
Oggier, M. et al. Second-year sea-ice salinity, temperature, density, oxygen and hydrogen isotope composition from the main coring site (MCS-SYI) during MOSAiC legs 1 to 4 in 2019/2020. PANGAEA https://doi.org/10.1594/PANGAEA.959830 (2023).
Lei, R. et al. Seasonality and timing of sea ice mass balance and heat fluxes in the Arctic transpolar drift during 2019–2020. Elementa: Science of the Anthropocene 10, 000089 (2022).
Macfarlane, A. R. et al. A Database of Snow on Sea Ice in the Central Arctic Collected during the MOSAiC expedition. Sci Data 10, 398 (2023).
Clum, A. et al. DOE JGI Metagenome Workflow. mSystems 6, https://doi.org/10.1128/msystems.00804-20 (2021).
Bankevich, A. et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology 19, 455–477 (2012).
Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner (2014).
Kang, D. et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35, 1026–1028 (2017).
Keeling, P. J. et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the Functional Diversity of Eukaryotic Life in the Oceans through Transcriptome Sequencing. PLoS Biol 12, e1001889 (2014).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–745 (2016).
Hofmeyr, S. et al. Terabase-scale metagenome coassembly with MetaHipMer. Sci Rep 10, 10689 (2020).
Saary, P., Mitchell, A. L. & Finn, R. D. Estimating the quality of eukaryotic genomes recovered from metagenomic analysis with EukCC. Genome Biol 21, 244 (2020).
Alexander, H. et al. Eukaryotic genomes from a global metagenomic data set illuminate trophic modes and biogeography of ocean plankton. mBio 14, e01676–23 (2023).
Lukashin, A. V. & Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Research 26, 1107–1115 (1998).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A. & Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res 41, e121 (2013).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Research 49, D412–D419 (2021).
Haft, D. H. et al. TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res 29, 41–43 (2001).
Sillitoe, I. et al. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res 41, D490–D498 (2013).
Wilson, D. et al. SUPERFAMILY–sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res 37, D380–386 (2009).
Letunic, I., Doerks, T. & Bork, P. SMART 6: recent updates and new developments. Nucleic Acids Res 37, D229–232 (2009).
Bland, C. et al. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 209 (2007).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol 1962, 1–14 (2019).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet 25, 25–29 (2000).
The Gene Ontology Consortium. et al. The Gene Ontology knowledgebase in 2023. Genetics 224, iyad031 (2023).
Levy Karin, E., Mirdita, M. & Söding, J. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 8, 48 (2020).
Grigoriev, I. V. et al. PhycoCosm, a comparative algal genomics resource. Nucleic Acids Research 49, D1004–D1011 (2021).
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B. & Wu, C. H. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res 33, W116–W120 (2005).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654 (2021).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res 10, 516–522 (2000).
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30, 1575–1584 (2002).
Boulton, W. et al. Metagenome-assembled-genomes recovered from the Arctic drift expedition MOSAiC. Figshare https://doi.org/10.6084/m9.figshare.27879576 (2024).
NCBI BioProject http://identifiers.org/ncbi/bioproject:PRJNA1160706 (2024).
Mukherjee, S. et al. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Research 49, D723–D733 (2021).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497145 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497146 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497147 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497148 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497149 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497150 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497151 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497152 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497153 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497154 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497155 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497157 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497160 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497161 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497165 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497166 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497167 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497168 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497174 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497178 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497181 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497183 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497184 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497185 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497186 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497187 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497189 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497190 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497191 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497192 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497193 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497194 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497195 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497196 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497197 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497198 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497199 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497200 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497201 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497202 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497203 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497204 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497205 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497207 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497209 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497213 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497214 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497216 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP497220 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506343 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506344 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506347 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506348 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506350 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506351 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506352 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506353 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506355 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506356 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506357 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506359 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506366 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506368 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506369 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506371 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506373 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506374 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506375 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506376 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506379 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506382 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506389 (2024).
NCBI sequence read archive http://identifiers.org/insdc.sra:SRP506779 (2024).
Nixdorf, U. et al. MOSAiC Extended Acknowledgement (2021).
Acknowledgements
W.B. was supported by the Natural Environment Research Council and ARIES DTP [grant number NE/S007334/1]. A.T., V.M. and T.M. were supported by the Natural Environment Research Council grant [NE/W005654/1]. Data used in this manuscript were produced as part of the international Multidisciplinary drifting Observatory for the Study of the Arctic Climate (MOSAiC) with the tag MOSAiC20192020 (expedition AWI_PS_122_00). We thank all land and ship based contributors of the MOSAiC campaign130. This work was also supported through the Research Council of Norway through project HAVOC (grant no 280292), and the National Science Foundation through grant OPP-1735862. The work (10.46936/10.25585/60001271) conducted by the US Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, was supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
W.B. drafted the manuscript and wrote code for the coassembly pipelines. W.B. and S.C. annotated the near complete eukaryotic MAG. A.S., I.V. G., S.C., K.B., R.R., K.L. provided bioinformatics support for all the JGI infrastructure including assembling, binning, and annotating all the individual samples, and bioinformatics support for the coassembly pipeline. K.B., I.V.G., T.M., K.M. coordinated and managed the metagenome sequencing project. A.F., C.J.M.H., A.T., K.M., O.M., J.G., M.O., collected and processed the samples. M.A.G., A.L. and G.B. coordinated and supervised the HAVOC project. O.M., J.G., S.L.E., K.O. performed the DNA extractions. T.M., V.M., R.L., A.T. supported and supervised the conceptualisation, writing and editing of the manuscript. All authors read, edited, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boulton, W., Salamov, A., Grigoriev, I.V. et al. Metagenome-assembled-genomes recovered from the Arctic drift expedition MOSAiC. Sci Data 12, 204 (2025). https://doi.org/10.1038/s41597-025-04525-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04525-8
