
Abstract
As a globally recognized predatory mite, Phytoseiulus persimilis Athias-Henriot is known for its highly effective control of pest mites. Also, as a model species in Phytoseiidae, P. persimilis possesses unique biological characteristics, such as the first offspring developing into a male with a strict sex sequence. However, the genetic mechanisms have not been fully unrevealed yet. To lay the groundwork for genetic research, we presented a high-quality chromosomal genome of P. persimilis with PacBio HiFi and Hi-C data. The total length of genome is 214.23 Mb, of which 190.48 (88.91%) is anchored on 4 chromosomes. The scaffold N50 is 57.95 Mb and the BUSCO (Benchmarking Universal Single-Copy Orthologs) completeness is 98.3%. Repeat elements comprise 27.59% (59.10 Mb) of the genome. The genome contained 15,847 predicted protein-coding genes, 12,344 of which were annotated for function. This high-quality genome of P. persimilis would allow us to explore the genetic mechanism underlying the biological characteristics of the Phytoseiidae species, and provide possibilities for the industrial optimization of commercial predatory mites in the future.
Similar content being viewed by others

Background & Summary
Phytoseiulus persimilis Athias-Henriot (Acari, Phytoseiidae) stands out as one of the most successful predatory mites in the world. Although extremely small body size, it’s one of the mainstays of integrated pest management for control of pest mites in the greenhouse. P. persimilis is the specialist predator of Tetranychus1,2,3, with strong predation ability4 to T. lintearius5 and T. turkestanit6. Although no single or compound eyes, it can locate prey rapidly and accurately by chemical cues generated by preys or plants induced by phytophagous species7,8. Fast development from egg to adult takes less than a week, and limb regeneration occurs during the early stage of development - from three pairs of legs in larval stage to four pairs after the protonymph stage. In addition, P. persimilis has a distinctive sex regulatory mechanism where the first three offspring are always male, female and female with a strict sex sequence9. A full understanding of these biological traits requires wide-scale exploration in model phytoseiid genomes. Whereas, the lack of high-quality genomic information impedes the advanced research of P. persimilis and other predatory mites10,11,12. In current study, we focus on the assembly and annotation of the high-quality reference genome of P. persimilis to enrich the genome resources of Phytoseiidae. It can pave a path to understand the genetic basis of prey recognition, limb regeneration and sex determination of phytoseiid species.
In this paper, we introduce the new chromosome-level genome of P. persimilis by the PacBio high-fidelity (HiFi) approach (Fig. 1). The finalized assembly spans 214.23 Mb, with a scaffold N50 of 57.95 Mb and a 98.3% completeness based on BUSCO evaluation. Annotation analysis uncovers the 27.59% repeat sequences and 15,847 protein-coding genes in the genome. This comprehensive genomic dataset serves as a valuable resource for further research on P. persimilis.

The workflow of this study. The panes with green, orange, light blue and blue represent the processes of genome assembly, genome size estimation, transcript assembly and genome annotation, respectively.
Methods
Sample collection and genomic DNA sequencing
The colony of P. persimilis has been cultivated for over a decade at the Laboratory of Predatory Mites, Institute of Plant Protection, Chinese Academy of Agricultural Sciences in Beijing, China. P. persimilis was nurtured by cultivating bean seedlings (Phaseolus vulgaris L.) and establishing a predation system involving bean seedlings-T. urticae-P. persimilis. They all were maintained at 25 ± 1 °C, with a humidity level of 70 ± 5% and a photoperiod of L16:D813.
The total genomic DNA was extracted from a collective sample of 800 male and female adult specimens, utilizing the Qiagen DNeasy Blood & Tissue Kit (Germany). Prior to extraction, the specimens were washed by sodium hypochlorite for 3 min to eliminate surface contaminants. For genome survey analysis, we created Illumina short-read DNA libraries (150 bp paired-end, 19.2 Gb, ~90X). A long-read DNA library was constructed from over 5 μg of DNA solution and sequenced on the PacBio Sequel II platform at GrandOmics, Beijing, China (44 Gb, ~205X). To aid in protein-coding gene prediction, total RNA was extracted from various developmental stages of P. persimilis, including egg, larval, protonymph, deutonymph, female adult, and male adult samples. Subsequently, short-read RNA libraries were prepared and sequenced on the Illumina platform (150 bp paired-end, 68.95 Gb).
Genome assembly
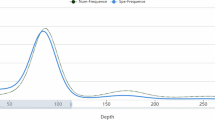
The genome size of P. persimilis was determined through k-mer analyses utilizing raw Illumina short-reads and raw PacBio HiFi reads. A k-mer distribution (k = 21) was created using Jellyfish v2.3.114, and the genome size was estimated utilizing Genoscope v1.0.015. Following the 21-mer depth analysis, resulting in an approximate size of 190 Mb with 1.3% heterozygosity (Fig. 2). The assembly of the P. persimilis genome was accomplished using PacBio HiFi reads with Hifiasm v0.19.5-r59316 under default settings, resulting in a draft genome of 228.49 Mb. It comprised 520 contigs, with an N50 length of 59.68 Mb and the largest contig of 63.92 Mb (Table 1). To refine the primary assembly and eliminate redundancy, we employed purdge v1.2.617. Then we used blobtools v1.1.118 to identify and remove bacterial contamination sequences. The accuracy and completeness of the assembly were evaluated using (i) QUAST v5.0.219 and (ii) BUSCO v5.4.720, based on the Arachnia_odb10 lineage data (https://busco-data.ezlab.org/v5/data/lineages/).

Estimation of P. persimilis genome size with 21-mer based on raw Illumina short-reads (a) and raw PacBio HiFi reads (b).
The assembly size closely aligned with the estimated genome size of approximately 190 Mb derived from k-mer analysis. The assembly genome exhibited a high level of completeness, reaching to 98.3%. Out of the 2,934 genes in Arachnia BUSCOs database, 94.6% were identified as complete and single-copy, 3.7% were complete but duplicated, 0.6% were fragmented, and 1.1% were missing (Table 2).
Hi-C scaffolding
We analyzed the Hi-C reads (accession number: SRR2341037721) and performed quality control by removing low-quality reads and adapters utilizing Trimmomatic v0.3922 (settings: ILLUMINACLIP:TruSeq. 3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36). The clean data were then aligned to the contig assembly utilizing HiC-Pro v3.1.023 to identify invalid pairs, and Yahs v1.124 was utilized to establish interactions. The assembly was manually reviewed using Juicebox v1.11.0825.
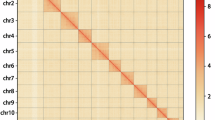
In summary, the chromosome-level genome exhibited a size of 190.48 Mb with a scaffold N50 of 57.95 Mb (Table 1). Approximately 88.91% (190.48 Mb) of the bases were successfully anchored onto four pseudo-chromosomes (Fig. 3), which was consistent with the results of karyotypes studies26.

Genome-wide Hi-C data heatmap and circular chromosome representation in P. persimilis genome assembly. (a) The heatmap of chromosome interactions. Colors indicate the frequency of Hi-C links, ranging from yellow (low) to red (high). The black box represents a chromosome. (b) Circos plot of the genomic elements distribution. The tracks indicate (i) length of the chromosome, (ii) gene density, colors indicate ranging from orange (low) to red (high) (iii) GC density, colors indicate the density of GC ranging from light green (low) to dark green (high), and (iv) density distribution of transposable element (TE). The densities of genes, GC, and TEs were calculated in 100 kb windows. Center: photo of a P. persimilis female adult.
Repetitive elements and protein-coding genes annotation
A de novo repeat library was constructed using RepeatModeler v1.0.1127. Subsequently, RepeatMasker v4.1.228 was employed with the filtered de novo repeat library to identify soft-mask repeats in the draft assembly before annotation. In total, 59.10 Mb of repetitive sequences, representing 27.59% of the total assembled genome, were detected. The major repeat elements included SINEs (0.01%), LINEs (2.41%), LTR elements (2.11%), DNA elements (4.65%), Rolling-circles (0.45%), Satellites (0.10%), and Unclassified (16.70%) (Table 3).
Following the masking of repeat sequences, a comprehensive annotation pipeline was implemented involving three key strategies: transcriptome-based prediction, homology-based prediction, and ab initio prediction for protein-coding genes. For transcriptome-based prediction, Trinity v2.15.129,30 was utilized to assemble the transcriptome and then predicted protein-coding genes with PASA v2.5.331,32,33,34. Homology-based prediction employed Miniport v0.1335, comparing gene structures with the Arthropoda protein dataset from OrthoDBv11(Bioinformatics Web Server - University of Greifswald, uni-greifswald.de). Ab initio prediction was performed using BRAKER v3.0336,37,38,39,40,41,42,43,44,45,46,47 to generate gene models based on short-read RNA-seq transcriptome data. Subsequently, EVidenceModeler v2.1.032,34 integrated the results from the three strategies to produce a non-redundant gene set, assigning weights as follows: PROTEIN: 3; TRANSCRIPT: 10; ABINITIO_PREDICTION:1. Overall, 15,905 genes were obtained, and 15, 847of the genes were protein coding genes (Table 4). The average gene length was 15,868.01 bp. The mean number of exons, introns, and CDS of each gene were 11.5, 9.8, and 10.6, respectively. The average length of exons, introns, and CDS of each gene were 472.33 bp, 877.58, and 255.34 bp, respectively.
Gene functional annotation was carried out by aligning protein sequences were aligned with Non-Redundant protein (NR), Universal Protein (UniProt), and Protein Families Analysis and Modeling (Pfam) databases. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) libraries were also utilized through GhostKOALA (kegg.jp)48 and PANNZER2 (helsinki.fi)49, respectively. Consequently, 12,344 protein-coding genes were successfully annotated for their functions (Table 5).
Data Records
The WGS, RNA seq, and PacBio HiFi data for the P. persimilis genome can be found on NCBI with the accession numbers SRR2967425150, SRR29686341-SRR2968634651,52,53,54,55,56, SRR2968644157 respectively under BioProject accession number PRJNA1128535. The genome data also can be found on NCBI (accession number: JBKEIU00000000058) and Figshare59.
Technical Validation
We assessed the quality of the chromosome-level genome of P. persimilis based on three key aspects: continuity, consistency, and completeness. For assessing the continuity of the genome, we determined that the scaffold N50 for P. persimilis is 57.95 Mb (Table 1). For evaluating the consistency of the genome, we analyzed the alignment of Illumina short-reads utilizing BWA 0.7.17-r118860, which indicated that 98.3% of the short reads were successfully aligned to the reference genome. To gauge the completeness of the chromosome-level genome, we utilized BUSCO v5.4.720,61 and referenced the 2,934 genes in Arachnia_odb10. The results demonstrated a high level of completeness, with percentages of 98.3%, 98.4% (Tables 2), and 96.1% for complete genes identified at the contig-level genome, chromosome-level genome, and protein level, respectively. These assessments collectively demonstrate the robust quality of the chromosome-level genome of P. persimilis.
Code availability
All software and pipelines were executed according to the manual and protocols of the published bioinformatic tools. The version and code/parameters of software have been described in Methods section. No custom code was used.
References
McMurtry, J. A., Moraes, G. J. D. & Sourassou, N. F. Revision of the lifestyles of Phytoseiid mites (Acari: Phytoseiidae) and implications for biological control strategies. Syst Appl Acarol. 18, 297–321 (2013).
Cruz-Miralles, J. et al. Plant-feeding may explain why the generalist predator Euseius stipulates does better on less defended citrus plants but Tetranychus-specialists Neoseiulus californicus and Phytoseiulus persimilis do not. Exp Appl Acarol. 83, 167–182 (2021).
Walzer, A., Paulus, H. F. & Schausberger, P. Ontogenetic shifts in intraguild predation on thrips by phytoseiid mites: the relevance of body size and diet specialization. Bull Entomol Res. 94, 577–584 (2004).
Skirvin, D. J. & Fenlon, J. S. Plant species modifies the functional response of Phytoseiulus persimilis (Acari: Phytoseiidae) to Tetranychus urticae (Acari: Tetranychidae): implications for biological control. Bull Entomol Res. 91, 61–67 (2001).
Davies, J. T., Ireson, J. E. & Allen, G. R. Pre-adult development of Phytoseiulus persimilis on diets of Tetranychus urticae and Tetranychus lintearius: implications for the biological control of Ulex europaeus. Exp Appl Acarol. 47, 133–145 (2009).
Wang, Y., Tuerxun, H. J. & Guo, W. C. The life table of Phytoseiulus persimilis fed on Tetranychus turkestani. Chin J Bio Con. 30, 329–333 (2014).
Azandémè-Hounmalon, G. Y. et al. Visual, vibratory, and olfactory cues affect interactions between the red spider mite Tetranychus evansi and its predator Phytoseiulus longipes. J Pest Sci. 89, 137–152 (2016).
Anja Dieleman, J., Marjolein Kruidhof, H., Weerheim, K. & Leiss, K. LED lighting strategies affect physiology and resilience to pathogens and pests in eggplant (Solanum melongena L.). Front Plant Sci. 11, 610046 (2021).
Zhang, B., Wang, E., Lv, J. & Xu, X. Impact of Gamma irradiation on reproduction and sex determination in Phytoseiulus persimilis Athias-Henriot. Chin J Bio Con. 6, 681–688 (2016).
Zhang, Y. X. et al. Genomic insights into mite phylogeny, fitness, development, and reproduction. BMC Genomics. 20, 954 (2019).
Hoy, M. A. et al. Genome sequencing of the Phytoseiid predatory mite Metaseiulus occidentalis reveals completely atomized hox genes and superdynamic intron evolution. Genome Biol Evol. 8, 1762–1775 (2016).
Jeyaprakash, A. & Hoy, M. A. The nuclear genome of the phytoseiid Metaseiulus occidentalis (Acari: Phytoseiidae) is among the smallest known in arthropods. Exp Appl Acarol. 47, 263–273 (2009).
Yan, H., Wang, E. D., Wei, G. S., Zhang, B. & Xu, X. N. Both host and diet shape bacterial communities of predatory mites. Insect Sci. 31, 551–561 (2024).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Laetsch, D. R. & Blaxter, M. L. BlobTools: interrogation of genome assemblies. F1000Research. 6, 1287 (2017).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29, 1072–1075 (2013).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of Eukaryotic, Prokaryotic, and Viral genomes. Mol Biol Evol. 38, 4647–4654 (2021).
NCBI Sequence Archive https://identifiers.org/ncbi/insdc.sra:SRR23410377 (2024).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics. 39, btac808 (2023).
Durand, N. C. et al. Juicebox Provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Wysoki, M. & McMurtry, J. A. Karyotypes of eight species of Phytoseiid mites of the genus Amblyseius Berlese (Acarina: Mesostigmata). Genetica. 47, 237–239 (1977).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA. 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 4, 4.10.1–4.10.14 (2009).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 8, 1494–1512 (2013).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Rhind, N. et al. Comparative functional genomics of the fission yeasts. Science. 332, 930–936 (2011).
Haas, B. J., Zeng, Q., Pearson, M. D., Cuomo, C. A. & Wortman, J. R. Approaches to Fungal Genome Annotation. Mycology. 2, 118–141 (2011).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics. 39, btad014 (2023).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 24, 637–644 (2008).
Stanke, M., Schöffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 7, 62 (2006).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinform. 22, 566 (2021).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-genome annotation with BRAKER. Methods Mol Biol. 1962, 65–95 (2019).
Hoff, K. J., Lange, S., Lomsadze, A., Borodovsky, M. & Stanke, M. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 32, 767–769 (2016).
Lomsadze, A., Paul, D. B. & Mark, B. Integration of mapped Rna-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119 (2014).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P. & Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 27, 1691–1692 (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 37, 907–915 (2019).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 1962, 161–177 (2019).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinform. 19, 189 (2018).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 428, 726–731 (2016).
Törönen, P. & Holm, L. PANNZER- a practical tool for protein function prediction. Protein Sci. 31, 118–128 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29674251 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29686341 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29686342 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29686343 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29686344 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29686345 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29686346 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29686441 (2024).
Xinyuan, Z. Phytoseiulus persimilis genome. GenBank https://identifiers.org/ncbi/insdc:JBKEIU000000000 (2025).
Zhou, X. Y. Genome assembly file of Phytoseiulus persimilis. figshare https://doi.org/10.6084/m9.figshare.27673563 (2024).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://doi.org/10.48550/arXiv.1303.3997 (2013).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol. 1962, 227–245 (2019).
NCBI Phytoseiulus persimilis Genome https://identifiers.org/ncbi/insdc.gca:GCA_037576195.1 (2024).
NCBI Metaseiulus occidentalis Genome https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000255335.2/ (2012).
NCBI Varroa destructor Genome https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_002443255.2/ (2017).
NCBI Varroa jacobsoni Genome https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_002532875.2/ (2017).
NCBI Tropilaelaps mercedesae Genome https://identifiers.org/ncbi/insdc.gca:GCA_002081605.1 (2017).
Acknowledgements
This work was supported by National Key R&D Program of China (2023YFD1400600). We thank Dr. Yiyuan Li (Institute of Plant Virology, Ningbo University, China) for technical advice on genome assemble and annotation and Dr. Bingyan Li (College of Plant Protection, China Agricultural University, China) for technical advice and manuscript revision. We also appreciate Ms. Hong Yan for mite collection and sample extraction.
Author information
Authors and Affiliations
Contributions
B.Z., X.X. and E.W. conceived and supervised the study. Z.X. performed the data analyses and wrote the manuscript. All authors have read, revised, and approved the final manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, X., Wang, E., Xu, X. et al. Chromosome-level genome assembly of Phytoseiulus persimilis Athias-Henriot. Sci Data 12, 293 (2025). https://doi.org/10.1038/s41597-025-04631-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04631-7
