
Abstract
Durian (Durio zibethinus) is a tropical fruit valued for its nutritional and commercial significance. In this study, we generated two high-quality, haplotype-resolved, chromosome-level genomes of the durian cultivar ‘Kan Yao’, each with 28 chromosomes. The total genome lengths were 737.2 Mb and 763.8 Mb, with contig N50 values of 22.9 Mb and 21.5 Mb, and scaffold N50 values of 25.9 Mb and 26.7 Mb, respectively. Nineteen chromosomes were assembled without gaps, while the remaining nine contained 1 to 10 gaps. Genome annotation identified 53,125 and 53,101 functional genes, as well as 5,254 and 5,496 non-coding RNAs. The high-quality assembled genomes will aid in the molecular breeding of durian.
Similar content being viewed by others

Background & Summary
The durian (Durio zibethinus), a tropical fruit tree belonging to the Malvaceae family, is often hailed as the “king of tropical fruits” due to its strong aroma and unique flavor1,2. Native to the Malay Archipelago, renowned for its unique flavor and rich nutritional value, it enjoys widespread popularity globally, boasting high nutritional and commercial value3,4,5. The durian has a hard outer shell covered with spines, while its flesh consists of segments enclosed within a leathery pericarp. The flesh is pale yellow, sticky, and juicy, with a distinctive aroma that elicits mixed opinions. They typically thrive in lowland tropical rainforests, requiring ample rainfall and sunlight.
In recent years, there have been some advancements in the study of the durian genome. In 2017, the full genome sequencing of the durian variety ‘Musang King’ was completed for the first time6. The size of the durian genome is approximately 750 Mb, with a chromosome number of 2n = 56. The challenge in studying the durian genome primarily stems from its complexity, as it contains a large number of repetitive sequences, increasing the difficulty of genome assembly6. However, single-molecule real-time sequencing technologies such as PacBio and Oxford Nanopore can assist in assembling longer and more complete chromosome-level genomes7,8,9,10. There may be considerable genetic variations among different durian varieties, which could be associated with various characteristics of durian such as size, flavor, color, and adaptability. Sequencing and comparing the genomes of multiple durian varieties can help reveal genetic variations and effects of these genetic variations.
Following the study workflow depicted in Fig. 1, we initially conducted next-generation sequencing (Illumina), PacBio HiFi, Nanopore ultralong (ONT), and High-throughput Chromosome Conformation Capture (Hi-C). These techniques were utilized to assemble contiguous and complete chromosome-scale haploid genomes of the ‘KanYao’ durian. Subsequently, structural gene annotation was performed using transcriptome data and homologous protein sequence information, along with the prediction of non-coding genes. Finally, functional annotation of the structural genes was carried out using public databases. This research provides essential foundational data and a theoretical basis for understanding the genetic characteristics of durian.

Schematic overview of the study design for durian genome sequencing and analysis, illustrating the key steps involved, including library preparation, sequencing technologies used (HiFi, ONT, and Hi-C), data processing, assembly, and annotation.
Methods
Sample preparation
The samples were collected from the germplasm nursery of durian in Baoting Li Autonomous County, Sanya City, Hainan Province, China. Young leaves were used for Illumina, HiFi, ONT, and HiC library construction and sequencing. Samples including stem, leaf, flower, seed, fruit at 7 days after flowering (fruit_PFD_7d), fruit pulp at 30 days after flowering (fruit_PFD_30d_pulp), fruit pulp at 60 days after flowering (fruit_PFD_60d_pulp), fruit pulp at 120 days after flowering (fruit_PFD_120d_pulp), fruit pulp at 150 days after flowering (fruit_PFD_150d_pulp), fruit stalk at 30 days after flowering (fruit_PFD_30d_stalk), fruit stalk at 60 days after flowering (fruit_PFD_60d_stalk), fruit spongy layer at 30 days after flowering (fruit_PFD_30d_SL), and fruit spongy layer at 60 days after flowering (fruit_PFD_60d_SL) were collected for RNA-seq and Iso-Seq library construction and sequencing.
Nucleic acid extraction
DNA extraction was performed using the Plant Genomic DNA Extraction Kit (Tiangen, DP320-03), while RNA extraction utilized the Plant Total RNA Extraction Kit (Tiangen, DP432). The integrity was assessed through 1% agarose gel electrophoresis, purity was determined using Nanodrop, and nucleic acid concentration was measured using the Qubit® DNA Assay Kit in the Qubit® 3.0 Fluorometer (Invitrogen, USA).
Library construction and sequencing
Illumina library construction and sequencing protocol: A total amount of 0.2 μg DNA per sample was used for the DNA library preparations. The sequencing library was generated using the Rapid Plus DNA Lib Prep Kit for Illumina (RK20208) following the manufacturer’s recommendations and index codes were added to each sample. Briefly, genomic DNA sample was fragmented by sonication to a size of 350 bp. Then DNA fragments were endpolished, A-tailed, and ligated with the full-length adapter for Illumina sequencing, followed by further PCR amplification. After purifying PCR products with the AMPure XP system (Beckman Coulter, Beverly, USA), DNA concentration was measured using the Qubit®3.0 Fluorometer (Invitrogen, USA). Libraries were then analyzed for size distribution with the Agilent 2100 Bioanalyzer and quantified by real-time PCR (>2 nM). The clustering of the index-coded samples was carried out on a cBot Cluster Generation System using Illumina PE Cluster Kit (Illumina, USA) following the manufacturer’s instructions. After cluster generation, the DNA libraries were sequenced on the Illumina HiSeq. 2000 platform and 150 bp paired-end reads were generated.
PacBio (CCS) library construction and sequencing protocol: Genomic DNA (gDNA) was sheared using a g-TUBE to fragments between 6 and 20 kb for 10 kb and 20 kb SMRTbell library construction. DNA with long overhangs was treated with ExoVII before damage repair. Repair enzymes from the Template Prep Kit were utilized to correct various DNA damages. T4 DNA Polymerase filled in 5’ overhangs and removed 3’ overhangs, while T4 PNK phosphorylated 5’ ends. Hairpin adapters were ligated to the repaired ends. Excess and imperfect SMRTbells were removed with ExoIII and ExoVII. AMPure PB Beads purified the library, which was further size-selected using the BluePippin System for large fragments. The selected SMRTbell templates were then purified again with AMPure PB Beads. A sequencing primer was attached, and a polymerase bound to the templates for loading into Zero-Mode Waveguides (ZWMs) using the Binding Kit. Finally, libraries were loaded into SMRT Cells, and sequencing was performed using the Sequel II instrument with the SMRT Cell 8 M Tray. The total length of the raw sequencing data is 95.7 Gb, with a total of 5,965,883 reads. The maximum read length is 58,876 bp, the minimum read length is 86 bp, the average read length is 16,038 bp, and the N50 is 19,782 bp.
ONT library construction and sequencing protocol: Genomic DNA was fragmented using a Covaris g-TUBE, followed by size verification. The DNA underwent repair and end-preparation with NEBNext End repair/dA-tailing Module and FFPE DNA Repair Mix, followed by purification with AMPure XP beads. Adapters from the NEBNext Quick Ligation Module were then ligated to the prepared DNA, followed by another round of AMPure XP bead cleaning. Specific fragment lengths were targeted using L or S Fragment Buffers. Finally, the DNA was eluted in Elution Buffer. The library prepared for sequencing was loaded onto the Oxford_Nanopore PromethION platform. The total length of the raw sequencing data is 94.4 Gb, with a total of 4,071,912 reads. The maximum read length is 718,956 bp, the minimum read length is 9 bp, the average read length is 23,178 bp, and the N50 is 53,343 bp.
HiC library construction and sequencing protocol: Hi-C libraries were constructed using an established protocol with modifications. The sample was ground in liquid nitrogen, cross-linked with 4% formaldehyde under vacuum at room temperature for 30 minutes, and the reaction was quenched with 2.5 M glycine. After chilling on ice, the sample was centrifuged, the pellet was washed with PBS, and subsequently, resuspended in lysis buffer. The supernatant was removed post-centrifugation. The nuclei were washed with NEB buffer, resuspended, and solubilized with SDS, then incubated at 65 °C. Triton X-100 was used to quench the SDS. An overnight digestion with DpnII was performed at 37 °C. DNA ends were marked with biotin-14-dCTP, and blunt-end ligation was conducted. The chromatin was ligated and cross-links were reversed by proteinase K treatment at 65 °C. DNA purification followed using phenol-chloroform extraction. Biotin from unligated ends was removed with T4 DNA polymerase. Sonication sheared DNA ends were repaired, and biotin-labeled Hi-C samples were enriched with streptavidin beads. A-tails were added, Illumina PE adapters were ligated, and the libraries were PCR-amplified before being sequenced on an Illumina HiSeq. 2000 platform.
RNA-seq library construction and sequencing protocol: Sequencing libraries were generated using the NEBNext Ultra RNA Library Prep Kit for Illumina following the manufacturer’s protocol, and index codes were assigned to each sample. mRNA was isolated from total RNA with poly-T magnetic beads and fragmented using divalent cations at high temperatures. First-strand cDNA synthesis was conducted using random primers and M-MuLV Reverse Transcriptase, followed by second-strand synthesis with DNA Polymerase I and RNase H. Overhangs were blunted, and 3’ ends were adenylated. NEB Next Adaptors with hairpin loop structures were ligated to the cDNA. The library fragments were then size-selected to 370-420 bp using the AMPure XP system. USER Enzyme was applied to adaptor-ligated cDNA, followed by PCR with Phusion High-Fidelity DNA polymerase and primers. PCR products were purified and the library quality was assessed on the Agilent 5400 system and quantified by QPCR. The qualified libraries were pooled and sequenced on an Illumina HiSeq. 2000 platform using a PE150 approach by Novogene Bioinformatics Technology Co., Ltd. Each sample undergoes independent library construction with three biological replicates.
Iso-Seq library construction and sequencing protocol: Iso-Seq libraries were prepared with the SMRTbell® Prep Kit 3.0 according to the prescribed method. Total RNA was isolated and mRNA was enriched. The mRNA was then reverse-transcribed into cDNA using an oligo-dT primer. Double-stranded cDNA was synthesized and subjected to damage repair and end-conditioning to create blunt-ended, 5’-phosphorylated ends. SMRTbell adapters were ligated to the prepared cDNA, forming the SMRTbell template required for sequencing. Exonuclease treatment was applied to remove failed ligation products and to ensure that only SMRTbell templates were present. The library was then size-selected to the desired range using the BluePippin™ Size Selection System. Following size selection, the library was assessed for quality and quantity. Once validated, the library was sequenced on a PacBio Sequel II system, which employed Single Molecule, Real-Time (SMRT) technology to generate long reads for full-length transcript sequencing.
Whole-genome sequencing yielded a total of 106 Gb of Illumina reads (~140× coverage), 95.7 Gb of HiFi reads (~125× coverage), 75.3 Gb of ONT reads (~100× coverage), and 90.3 Gb of Hi-C reads (~120× coverage). Illumina reads and Hi-C reads were filtered with the default parameters of fastp (v0.20.0)11 software. HiFi and ONT reads were filtered using Filtlong (https://github.com/rrwick/Filtlong) software, retaining reads longer than 12 kb and 30 kb with quality scores above 90% to avoid possible errors, respectively. HiFi and ONT filtered reads of 86.1 Gb (~110× coverage) and 75.3 Gb (~96.5× coverage) were used for genome assembly.
Genome survey and analysis
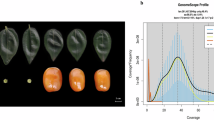
The clean ILLUMINA reads were then used for k-mer counting with Jellyfish (v2.3.0)12 software. Following 21-mer counting, the resulting matrix was utilized to calculate the haploid genome size and heterozygosity using Genomescope (v2.0)13 with parameters -p 2 -k 21. The results indicate that the genome size is approximately 753 Mb, with a high heterozygosity of around 1.23% (Fig. 2).

Genome survey results based on K-mer analysis. (a) Phenotypic images of ‘Kan Yao’ durian, including the plant, flower, whole fruit, and cross-section of the fruit. (b) Analysis of genome size and heterozygosity using Jellyfish (v2.3.0) and Genomescope (v2.0) with parameters -p 2 -k 21.
Assessment of sequencing data saturation
HiFi reads were randomly divided into different coverage levels (10×, 30×, 50×, 70×, 90×, 110×), and initial assemblies were performed with Hifiasm (v0.19.5)14 software to evaluate data saturation. The results indicate that with the increase in sequencing data, the total length of the assembled sequences also increases (Fig. 3). However, the contig N50 reaches a plateau at 70× coverage, while BUSCO (v4.1.4) completeness plateaus at 30× coverage. Consequently, the HiFi sequencing data in this study (~110×) have achieved saturation, with the optimal coverage being 70×.

Evaluation of Assembly Metrics at Different Sequencing Data Coverage Levels. (a) Line graph showing the contig N50 values for assemblies at various coverage levels (10×, 30×, 50×, 70×, 90×, 110×). (b) Evaluation of total sequence length and BUSCO completeness for assemblies at different coverage levels (10×, 30×, 50×, 70×, 90×, 110×).
Genome assembly
Genome contigs were assembled using Hifiasm (v0.19.5)14 software, incorporating HiFi, ONT, and Hi-C reads (hifiasm -o asm -t48–h1 hic_clean_1.fq.gz–h2 hic_clean_2.fq.gz hifi.fastq.gz–ul ONT.fastq.gz), resulting in hap1 and hap2 haplotype genome drafts. Chromosome mounting and assembly of contigs was conducted using 3D-DNA (v190716)15, followed by manual correction with Juicebox (v1.11.08)16 software. Twenty-eight chromosomes were extracted and used as a reference genome for quarTeT AssemblyMapper (v1.0.3)17 software to re-anchor the haplotype data from Hifiasm. ONT data were then used to fill the gaps by quarTeT GapFiller (v1.0.3)17, resulting in the final genome assemblies, designated as hap1 and hap2, respectively. 21 chromosomes of hap1 had no gaps, whereas 22 chromosomes of hap2 had no gaps (Fig. 3). The assembly results were analyzed using QUAST (v5.0.2)18 software. The total genome sizes for hap1 and hap2 were 737.2 Mb and 763.8 Mb respectively, closely aligning with the estimated size of ~753 Mb. The contig N50 values were 22.9 Mb and 21.5 Mb, while the scaffold N50 values were 25.9 Mb and 26.7 Mb, respectively (Table 1). Telomere detection was performed using TIDK software (https://github.com/tolkit/telomeric-identifier), identifying the telomeric repeat unit as AAACCCT. All 28 chromosomes of the hap1 genome had telomeres detected at one or both ends (Fig. 4), with telomeres present at both ends of 23 chromosomes and at only one end of 5 chromosomes (chr7A, chr12A, chr20A, chr23A, chr25A). Similarly, all 28 chromosomes of the hap2 genome had telomeres detected at one or both ends. Telomeres present at both ends of 22 chromosomes and at only one end of 6 chromosomes (chr7B, chr11B, chr12B, chr20B, chr23B, chr25B). Our assembly results were also compared with a recently published genome of the Durio zibethinus ‘Kan Yao’ cultivar19, which only assembled a single haploid genome. In contrast, our study successfully assembled two haploid genomes. While both assemblies show similar results in terms of chromosome number, total length of sequences anchored to chromosomes, proportion of repetitive sequences, and BUSCO scores, our two haploid assemblies exhibit superior performance in terms of Contig N50 and QV values (Table 1).

Schematic diagram of chromosome structure. The upper diagram represents the hap1 haplotype genome, while the lower diagram represents the hap2 haplotype genome. Red triangles indicate telomeres, and purple lines represent gaps.
Genome annotation
Repeat sequence annotation and masking were performed using RepeatModeler (v2.0.1)20 and RepeatMasker (v4.0.7)21. First, the genome sequence was input into RepeatModeler, which identifies repeat sequence models based on the Dfam (v3.2)22 and RepBase-2018102623 databases to build a repeat sequence library. Then, RepeatMasker annotated the repeat sequences and converted them to lowercase letters. The proportion of repeat sequences in the hap1 and hap2 genomes was 56.75% and 58.48%, respectively. The types of repeat sequences included transposons such as DNA, LTR, LINE, SINE, and RC, as well as satellite DNA, simple repeats, and unknown types. The majority of repeat sequences were LTR transposons, specifically Gypsy (27.1% and 33.56%) and Copia (5.25% and 4.80%).
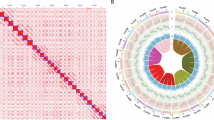
Gene structure annotation employed three strategies: de novo, homology-based, and transcriptome-based annotation. For de novo annotation, Braker (v2.1.6)24 was used to build models from Arabidopsis protein sequences (arabidopsis_pep_20101214.fa) and merged RNA-seq reads. Homology-based annotation was conducted with GenomeThreader (v1.7.3)25, referencing ‘Musang King’ durian protein annotations (GCF_002303985.1_Duzib1.0_protein.faa). Transcriptome-based annotation utilized PASA (v2.5.0)26 with Iso-Seq reads to accurately annotate the gene structures. The results from these strategies were then merged using EvidenceModeler (v.1.1.1)27 and updated with PASA to incorporate UTR and alternative splicing information, resulting in the final annotation file. The haplotype genomes hap1 and hap2 were annotated with 50,417 and 50,390 coding genes, corresponding to 92,276 and 91,712 transcripts, respectively (Table 2). Gene and repeat sequence densities were calculated using a 500 kb sliding window (bedtools makewindows -w 500000). Protein sequence homology within the genome was analyzed using DIAMOND (v2.0.4.142)28 software. Collinearity analysis was performed with MCScanX29, and visualizations were created using Circos (v0.69-8)30 (Fig. 5). BUSCO (v4.1.4)31 analysis comparing the CDS sequences of transcripts with the embryophyta_odb10 database (v2020-09-10) revealed that the two sets of haplotype genome contained approximately 94.1% and 94% complete homologous conserved genes, respectively (Table 2).

Circos plot of haplotype 1 (a) and haplotype 2 (b) genomes. The outer track represents the Gene density histogram, and the inner track represents the Repeat sequence density histogram.
The function annotation of the protein-coding genes followed this protocol: 1. DIAMOND (v2.0.4.142) was used with parameters -e 0.001 -f 5 -k 1 to align against the GenBank-NR (https://www.ncbi.nlm.nih.gov/protein), SwissProt32, and UniRef9033 databases; 2. The pfam_scan.pl script was run with parameters -clan_overlap -as -cpu 16 -e_seq. 1e-5 -e_dom 1e-5 to align against the Pfam34 database; 3. eggNOG-mapper (v2.0.0)35 was run with parameters –no_file_comments -m diamond to BLAST against the EggNOG (v5.0)36 database for GO and KO annotations. The annotation rates for the NR and UniRef90 databases were similar, both around 91%, while the rates for the Pfam and Swiss-Prot databases were lower, at approximately 70% (Table 2). Non-coding gene annotation was compared with the Rfam37 database using Infernal (v1.1.4)38 software, which identified 5,254 and 5,496 non-coding RNAs in hap1 and hap2, respectively. These included various types such as riboswitches, tRNAs, miRNAs, rRNAs, snRNAs, ribozymes, antisense RNAs, and sRNAs. The majority were rRNAs, followed by snRNAs and tRNAs. A total of 281 and 284 miRNAs were annotated in hap1 and hap2, respectively (Table 2).
Comparative of haplotype genomes
Synteny analysis of the two haplotype genomes was performed using the MUMmer (v4.0.0)39 software, which demonstrated that the two haplotype genomes are nearly identical (Fig. 6). Hi-C interaction heatmap analysis was carried out using the HiCPlotter40 software, which clearly showed strong intra-chromosomal interaction signals and notable interaction signals between homologous chromosomes, while inter-chromosomal interaction signals were weak. The gene collinearity analysis between the two haplotype genomes was performed using the TBtools (v2.096)41 software. This analysis revealed that the majority of genes exhibit conserved chromosomal distributions between the two haplotype genomes, while a small number of genes show evidence of rearrangements or translocations.

Comparative analysis of the two haplotype genome structures. (a) Genomic sequence collinearity analysis. (b) Hi-C interaction heatmap analysis. (c) Gene collinearity analysis. (d) Chromosomal structural variation (SV) analysis.
Filtered ONT sequencing reads were utilized for chromosomal structural variation (SV) analysis with the NGMLR(v0.2.7)42 and Sniffles(v1.0.11)43 software. In total, 25,572 structural variation sites (SVs) were detected, including eight types: deletion (DEL), insertion (INS), duplication (DUP), breakend (BND), inversion (INV), deletion/inversion (DEL/INV), inversion/duplication (INVDUP), and duplication/insertion (DUP/INS) (Fig. 6). Among these, DEL was the most frequent with 12,864 occurrences, followed by INS with 10,282 occurrences.
Data Records
The sequencing data (PacBio HiFi, Nanopore ultralong, Hi-C, next-generation and RNA-seq reads) generated in this study have been deposited in the Genome Sequence Archive in National Genomics Data Center (NGDC), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA016775)44 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. and NCBI Sequence Read Archive with accession number SRR29483665-SRR2948370445,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84 and SRR31130142-SRR3113015985,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102, under BioProject number PRJNA1126441. The chromosomal assembly and dataset of gene annotation have been deposited at DDBJ/ENA/GenBank under the accession PRJNA1181610103 and PRJNA1181611104, and also been deposited at Nation Agriculture Science Data Center (https://doi.org/10.57760/sciencedb.agriculture.00013)105 and figshare database (https://figshare.com/articles/dataset/_/26112652)106.
Technical Validation
We evaluated the genome assembly and annotation from three aspects: continuity, completeness, and accuracy. Both haplotype genomes exhibit high continuity, with Contig N50 values of 22.9 and 21.5 Mb, and Scaffold N50 values of 25.9 and 26.7 Mb, respectively. The saturation assessment of sequencing data also indicated that increasing the data volume would not significantly improve the Contig N50. Using the BUSCO (v4.1.4) software with the embryophyta_odb10 database, the genome sequences were found to match 99.2% ([S:75.8%,D:23.4%],F:0.2%,M:0.6%) and 99.1% ([S:75.5%,D:23.6%],F:0.2%,M:0.7%) of the genes in the database, respectively, and the annotated transcript sequences matched 94.1% ([S:48.1%,D:46.0%],F:1.9%,M:4.0%) and 94% ([S:49.3%,D:44.7%],F:1.5%,M:4.5%) of the genes, demonstrating a high degree of completeness in both genome assembly and annotation. The Hi-C interaction heatmap showed independence of signals for each chromosome, and the collinearity analyses of genome sequences and genes between the two haplotype genomes showed high consistency.
Code availability
The pipeline used for genome assembly and annotation is detailed and available at https://github.com/CAAS000JXH/Durian_genome_assembly_pipline.
References
Li, J. X., Schieberle, P. & Steinhaus, M. Characterization of the major odor-active compounds in Thai durian (Durio zibethinus L. ‘Monthong’) by aroma extract dilution analysis and headspace gas chromatography-olfactometry. J. Agric. Food Chem 60, 11253–11262 (2012).
Chin, S. T. et al. Analysis of volatile compounds from Malaysian durians (Durio zibethinus) using headspace SPME coupled to fast GC–MS. J. Food Compos. Anal 20, 31–44 (2007).
Charoenkiatkul, S., Thiyajai, P. & Judprasong, K. Nutrients and bioactive compounds in popular and indigenous durian (Durio zibethinus murr.). Food Chemistry 193, 181–186 (2016).
Ho, L. H. & Bhat, R. Exploring the potential nutraceutical values of durian (Durio zibethinus L.) -An exotic tropical fruit. Food Chemistry 168, 80–89 (2015).
Ketsa, S. Durian—Durio zibethinus. Exotic Fruits. 169–180 (2018).
Teh, B. T. et al. The draft genome of tropical fruit durian (Durio zibethinus). Nat Genet 49, 1633–1641 (2017).
Girollet, N. et al. De novo phased assembly of the Vitis riparia grape genome. Scientifc Data 6, 1–8, 10/ghdrm3 (2019).
Ko, S. R. et al. High-quality chromosome-level genome assembly of Nicotiana benthamiana. Sci Data 11, 386 (2024).
Liu, D. et al. Haplotype-resolved chromosomal-level genome assembly of Buzhaye (Microcos paniculata). Sci Data 10, 901 (2023).
Peng, H. et al. A haplotype-resolved genome assembly of Malus domestica ‘Red Fuji’. Sci Data 11, 592 (2024).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18, 170–175 (2021).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Lin, Y. Z. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Research 10, uhad127 (2023).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Li, W., Chen, X., Yu, J. & Zhu, Y. Upgraded durian genome reveals the role of chromosome reshuffling during ancestral karyotype evolution, lignin biosynthesis regulation, and stress tolerance. Science China Life Sciences 67, 1266–1279 (2024).
Smit, A. & Hubley, R. RepeatModeler-1.0.11. Institute for Systems Biology, http://www.repeatmasker.org/RepeatModeler/ (2019).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0, http://www.repeatmasker.org/RMDownload.html (2015).
Hubley, R. et al. Te Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D8–D89 (2016).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR genomics and bioinformatics 3, lqaa108 (2021).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Information and Software Technology 47, 965–978 (2005).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature methods 12, 59–60 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40, e49–e49 (2012).
Krzywinski, M. et al. Circos: an Information Aesthetic for Comparative Genomics. Genome Res 19, 1639–1645 (2009).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 31, 365–370 (2003).
Suzek, B. E. et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Griffiths-Jones, S. Rfam: annotating non-coding RNAs in complete genomes. Nucleic. Acids. Res. 33, D121–D124 (2004).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Delcher, A. L., Salzberg, S. L. & Phillippy, A. M. Using MUMmer to identify similar regions in large sequence sets. Current protocols in bioinformatics, 10.13. 11-10.13. 18 (2003).
Akdemir, K. C. & Chin, L. HiCPlotter integrates genomic data with interaction matrices. Genome Biology 16, 198 (2015).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742 (2023).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods 15, 461–468 (2018).
Page, A. J., Bielawski, J. P. & Mott, R. Sniffles: A tool for high-throughput detection of structural variations in single-molecule sequencing data. Bioinformatics 34, 763–770 (2018).
National Genomics Data Center (NGDC) Genome Sequence Archive https://ngdc.cncb.ac.cn/search/all?&q=CRA016775 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483665 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483666 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483667 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483668 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483669 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483670 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483671 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483672 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483673 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483674 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483675 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483676 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483677 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483678 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483679 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483680 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483681 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483682 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483683 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483684 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483685 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483686 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483687 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483688 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483689 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483690 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483691 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483692 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483693 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483694 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483695 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483696 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483697 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483698 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483699 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483700 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483701 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483702 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483703 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29483704 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130142 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130143 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130144 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130145 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130146 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130147 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130148 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130149 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130150 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130151 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130152 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130153 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130154 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130155 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130156 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130157 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130158 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31130159 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_047325155.1 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_047325235.1 (2024).
Ji, X. H. Chromosome-scale haploid genome sequence and annotation dataset of the durian cultivar ‘Kan Yao’ [DS/OL]. V2. Science Data Bank https://doi.org/10.57760/sciencedb.agriculture.00013 (2024).
Ji, X. H. Assembly and Annotation of the Haploid Genome of the KanYao Durian. figshare. Dataset. https://doi.org/10.6084/m9.figshare.26112652.v1 (2024).
Acknowledgements
This work was supported by the Nanfan Special Project of the Chinese Academy of Agricultural Sciences (Grant No. SWAQ09), Hainan Academy of Agricultural Sciences Program (HAAS2022JBGS01) and the Innovation Project of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2021-RIP-02).
Author information
Authors and Affiliations
Contributions
X.H.J., F.Z.L., D.J.Z., X.J.F. and H.B.W. contributed to the research design. D.J.Z. and S.H.X. collected the samples. X.H.J., M.S., X.D.W. and Y.W.Z. analyzed the data. X.H.J., Y.W.Z., F.Z.L., D.J.Z., X.J.F. and H.B.W. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, X., Zhong, Y., Zheng, D. et al. Chromosome-scale haploid genome assembly of Durio zibethinus KanYao. Sci Data 12, 384 (2025). https://doi.org/10.1038/s41597-025-04656-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04656-y
This article is cited by
-
Applications of omics in deciphering the unique traits of durian, the king of fruits
Plant Molecular Biology (2026)
-
Genomic resequencing reveals genetic diversity, population structure, and core collection of durian germplasm
Communications Biology (2025)
