
Abstract
The darkbarbel catfish (Pelteobagrus vachelli), a species of significant economic value in China’s aquaculture sector, is widely utilized in hybrid yellow catfish production due to its exceptional growth rate. The growth rate of male P. vachelli is significantly higher compared to females, making all-male breeding a promising market opportunity. Therefore, the analysis of the male P. vachelli genome provides crucial genetic information for hybrid breeding and all-male breeding. Utilizing PacBio Hifi long-read sequencing and Hi-C technologies, we present a high-quality, chromosome-level genome assembly for the male P. vachelli. The assembly covers 728.88 Mb with 99.92% of the sequence distributed across 26 chromosomes. The contig N50 is 5.60 Mb, and the scaffold N50 is 28.76 Mb. The completeness of the P. vachelli genome assembly is highlighted by a BUSCO score of 97.45%. The genome is estimated to encode 25,121 protein-coding genes, with 93.46% annotated functionally and a BUSCO score of 96.40%. Repeat elements constitute approximately 38.97% of the genome. This comprehensive genome assembly represents an invaluable resource for advancing hybrid breeding, comparative genomics, and evolutionary studies in catfish and related species.
Similar content being viewed by others


Background & Summary
The darkbarbel catfish (Pelteobagrus vachelli) belongs to the order Siluriformes, family Bagridae, and genus Pelteobagrus1. This species is the largest and fastest-growing within its genus, with individuals reaching up to 2 kg in weight and 50 cm in length2,3. Due to its flavorful meat, nutritional richness, minimal intermuscular bones, and high nutritional value, P. vachelli have been highly sought after by consumers and the market, making them one of the fastest-growing specialty fish species in pond aquaculture over the past decade4,5. The yellow catfish (Pelteobagrus fulvidraco), a closely related species within the same genus, is also an important aquaculture species in China, with a production of 622,651 tons in 2023. Although P. vachelli grows faster and attains a larger weight compared to P. fulvidraco, its flesh is less tender. Recent research indicates that hybrid yellow catfish (P. fulvidraco ♀ × P. vachelli ♂) demonstrate significant advantages in growth, survival rate, disease resistance, and transportability6. Consequently, hybrid yellow catfish have gradually become the main aquaculture species, leading to the development of the new variety “Huangyou No. 1”7. As a promising new aquaculture variety, the hybrid yellow catfish has a very positive market outlook8. However, the molecular mechanisms underlying the hybrid heterosis in these interspecific hybrid yellow catfish remain unclear. Further research utilizing multi-omics analyses and other techniques is necessary to elucidate the genetic mechanisms and gene regulation responsible for this hybrid heterosis.
Species within the Bagridae genus exhibit significant sexual dimorphism in growth, making the study of sex determination mechanisms highly significant9,10. To date, several species have successfully decoded their chromosomal-level genomes, including the Pelteobagrus fulvidraco11, Leiocassis longirostris12, Pseudobagrus ussuriensis13, Hemibagrus wyckioides14,15, Hemibagrus macropterus16, Hemibagrus guttatus17. These genome sequences provide a solid foundation for analyzing key economic traits, particularly in the investigation of sex determination mechanisms, and eventual application in monosex breeding18,19,20. Pelteobagrus vachelli shows pronounced differences in growth between sexes in both wild and farmed populations. One-year-old males grow approximately 50% faster than females under the same farming conditions, while two-year-old males grow 2 to 3 times faster than females2,21. Chinese researchers have developed a sex-specific molecular marker-assisted technique for producing all-male fish based on these growth differences, which has significantly increased the yield and economic benefits of the yellow catfish industry22,23. Aquaculture practices have also demonstrated that significant growth differences between sexes persist in hybrid yellow catfish, with two-year-old males growing about 50% faster than females7. This suggests that there is still a necessity and potential for all-male breeding in hybrid yellow catfish24. Consequently, analyzing the genomic information of male P. vachelli, developing male sex-specific molecular markers, and establishing rapid methods for identifying the genetic sex of P. vachelli are crucial for the all-male breeding of P. vachelli and hybrid yellow catfish. While previous studies have reported the female genome25,26, information on the male genome remains scarce.
In this study, we utilized short-read, PacBio HiFi long-read sequencing and Hi-C technology to generate a high-quality, chromosome-level assembly of the male Pelteobagrus vachelli genome. The development of this reference genome is expected to drive significant advancements in population genetics and the identification of functional genes linked to key economic traits in P. vachelli. Establishing this genomic foundation has the potential to facilitate hybrid breeding and all-male breeding programs for P. vachelli.
Methods
Sample collection and DNA extraction
A mature male Pelteobagrus vachelli was collected from the Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou, China. Muscle tissue from this specimen was used to extract DNA for whole-genome sequencing, including short-read and long-read sequencing, as well as Hi-C sequencing. All experiments were conducted in accordance with the recommendations of the Ethics Committee of the Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences. Genomic DNA was extracted from the muscle tissue using a Qiagen DNeasy Blood and Tissue Kit (Qiagen, USA), following the manufacturer’s instructions. The quality and concentration of the extracted DNA were assessed using a NanoDrop One spectrophotometer (Thermo Scientific, USA) and 1% agarose gel electrophoresis.
Genome sequencing
The extracted DNA was randomly sheared into approximately 350 bp fragments, and a short-read library was constructed using the MGIEasy Universal DNA Library Prep Set (MGI, China). Sequencing was performed on the MGISEQ T7 platform (MGI, China), producing 38.89 Gb of paired-end raw reads, each 150 bp in length (Table 1). For PacBio sequencing, we employed the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, USA) following PacBio’s standard protocol with insert sizes of 15 kb, and sequenced on the Pacific Biosciences Sequel II platform in CCS mode. This process yielded 31.22 Gb of HiFi data, with an average read length of 14.05 kb (Table 1). For Hi-C sequencing, approximately 1 g of muscle tissue from the male Pelteobagrus vachelli was dissected and processed using the GrandOmics Hi-C kit (DpnII restriction enzyme; GrandOmics, China) according to the manufacturer’s protocol. The Hi-C library was sequenced on the MGISEQ T7 platform (MGI, China), yielding 81.36 Gb of Hi-C read data (Table 1).
RNA extraction and transcriptome sequencing
To facilitate genome annotation, total RNA was extracted from twelve tissues, including the brain, liver, kidney, heart, muscle, spleen, skin, gill, swim bladder, intestine, blood, and testis. The RNA quality was assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). A cDNA library was constructed from the mixed RNA sample using the MGIEasy Universal DNA Library Prep Set (MGI, China), following the manufacturer’s protocol. This library was sequenced on the MGISEQ T7 platform (MGI, China) with a paired-end 150 bp layout, producing 32.47 Gb of transcriptome data (Table 1).
Genome size and heterozygosity estimation
The raw genome MGI data were primarily filtered using fastp v 0.23.227 (parameter: -q 15 -l 150) to remove low-quality reads and adaptor sequences. To estimate the genome size of the P. vachelli, a k-mer analysis was performed using 38.05 Gbclean reads. Initially, Jellyfish (v2.3.0)15 was used to calculate the frequency of 17-mers and generate a k-mer frequency table. Subsequently, GenomeScope (v2.0)28 was used to analyze the overall genomic properties. The preliminary genome survey estimates that P. vachelli has a genome size of approximately 594,436,389 bp, with 0.568% heterozygosity and 39.3% of the genome repeat content (Fig. 1).

The 17-mer frequency distribution analysis chart for the Pelteobagrus vachelli genome.
Genome assembly
The genome assembly was performed using Hifiasm (v0.19.5)29 with 2,222,451 HiFi reads and a total of 31.22 Gb of data, employing the default parameters. HiFi long reads were input into Hifiasm to generate primary assembly contig graphs. This process resulted in 824 contigs with a total length of 728.88 Mb, a maximum contig size of 17.96 Mb, and an N50 of 5.60 Mb (Table 2). Scaffolding was achieved using Juicer (v1.6)30 in conjunction with 3D-DNA (v201008)31. Initially, BWA (v0.7.17)32 was employed to index the contig-level genome, after which Juicer was utilized to identify restriction enzyme cutting sites. Clean Hi-C (paired-end) reads were mapped to the contigs using Juicer, and Hi-C-assisted initial chromosome assembly was performed with the 3D-DNA algorithm following standard protocols. Chromosome boundaries were refined, and scaffolds corrected using the manually operated Juicerbox (v1.11.08)33 module, resulting in the resolution of 26 chromosomes (Figs. 2, 3). The file modified by Juicebox was further revised and used as input for 3D-DNA for re-scaffolding on a per-chromosome basis. The final assembly comprised 32 scaffolds, with a maximum scaffold size of 45.05 Mb and an N50 size of 28.76 Mb (Tables 2, 3).

Hi-C contact map produced by 3D-DNA.

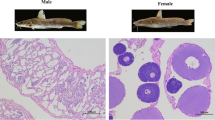
Features of the P. vachelli genome. From outside to inside: (a) The 26 pseudo-chromosomes, (b) GC content, (c) Gene density, (d) Repeats content, (e) LTR content, (f) LINE content, (g) DNA-TE content and (h) Male and female P. vachelli.
Repeat annotation
In recognition of the importance of tandem repeats, we employed two software tools, GMATA (v2.2.1)34 and Tandem Repeats Finder (TRF, v4.10.0)35, to perform a genome-wide search for tandem repeat sequences using default parameters. GMATA is designed primarily to identify simple sequence repeats (SSRs) with shorter repeat units, whereas TRF can detect tandem repeats of all types of repeat units. The search results indicated that SSRs comprise 1.49% of the total genome length, while tandem repeat sequences account for 1.99% of the genome length. We then investigated the dispersed repetitive sequences. Initially, MITE-hunter36 was used to identify a small transposon known as MITE within the genome, creating a MITE library file. Following this, a hard-masking procedure was applied to the genome, marking repeated sequences as ‘N’, and RepeatModeler (v2.05)37 was employed to conduct a de novo search for additional repeated sequences, resulting in the formation of a denovo library file (RepMod.lib). Given that RepMod.lib contained numerous unknown repeated sequences, TEclass38 was utilized for classification. Finally, the MITE.lib, RepMod.lib, and Repbase (v19.06)39 libraries were integrated to create a comprehensive library file. This total library file was then employed with RepeatMasker (v4.1.6)40 to conduct a search for repeated sequences throughout the entire genome. The results revealed that dispersed repetitive sequences constitute 32.53% of the total genome length (Table 4). Among transposable elements (TEs), DNA elements are the most prevalent, making up 15.21% of the genome, followed by long interspersed nuclear elements (LINEs) at 6.88%, long terminal repeat (LTR) retrotransposons at 5.00%, and miniature inverted repeat transposable element (MITE) at 2.92% of the genome. Ultimately, a total of 284,053,865 bp of repetitive sequences were identified, comprising 38.97% of the entire genome (Table 4, Fig. 4).

Four types of TE sequence divergence distribution diagram annotated by RepeatMasker.
Gene prediction and function assignment
Gene structure prediction was carried out using three distinct methodologies: homology-based, transcriptome-based, and ab initio annotations. For the homology-based prediction, we utilized GEMOMA (v1.6.1)41 to compare homologous proteins from six related species (Danio rerio, Ictalurus punctatus42, Silurus meridionalis43, Pangasianodon hypophthalmus44, Pseudobagrus ussuriensis13, and Pelteobagrus fulvidraco11) with our assembled genome. Transcriptome sequence annotation via PASA (v2.3.3)45 facilitated the acquisition of gene information. This information was then employed in a semi-supervised self-training process with GeneMark-ST46 (v5.1) to predict gene models. The predicted genes were compared against the Swissprot Database47 using Blastp, with alignment results filtered for identity ≥ 95%. We selected the top 3,000 genes with the highest alignment scores from GeneMark-ST as the training set for AUGUSTUS model training. Subsequently, AUGUSTUS (v3.5.0)48 was used to predict genes within the genome using the developed model. The gene prediction results from ab initio, homology-based, and transcriptome-based annotations were converted into a format compatible with EVM (v2.1.0)45. These files were then integrated using EVM with default parameters to produce an initial non-redundant gene set. Our predictions identified a total of 23,638 genes in the genome, with an average gene length of 14,706.84 bp, an average coding sequence length of 167.49 bp, and an average of 9.99 exons per gene (Table 5, Fig. 5).

Venn diagram of function annotations from various databases.
Data Records
The raw sequencing reads of all libraries have been deposited into NCBI SRA database via the accession number PRJNA100029449. The assembled genome has been deposited at Genbank under the accession number GCA_033026395.150. Moreover, data of the genome annotations, predicted coding sequences and protein sequences are available at Figshare51.
Technical Validation
Genome synteny analysis
To investigate chromosomal synteny with closely related species, we performed a comparative analysis using the genome of P. vachelli alongside those of Pelteobagrus fulvidraco11 and Pseudobagrus ussuriensis13. Whole genome DNA sequence alignments between P. vachelli and the other two species were conducted using MCscan (v0.8)52 and syntenic relationships were visualized with JCVI (v1.1.12)53. The collinearity analysis revealed chromosomal rearrangements on six chromosomes between P. vachelli and P. fulvidraco. However, the genomes of P. vachelli and P. ussuriensis exhibited a perfect one-to-one correspondence between their chromosomes, demonstrating the high quality and accuracy of our genome (Fig. 6).

A synteny analysis of the chromosomes among genomes of Pelteobagrus vachelli and the other two fish Pelteobagrus fulvidraco and Pseudobagrus ussuriensis.
Assessment of genome assembly
The accuracy of the P. vachelli genome assembly was evaluated by assessing its completeness using the conserved metazoan gene set ‘actinopterygii_odb10’ from BUSCO (v5.4.3)54. The analysis demonstrated high completeness, with an overall completeness of 98.1%. Specifically, 96.8% of the genes were complete and single-copy, 1.3% genes were complete and duplicated, 0.9% genes were fragmented, and 1.0% genes were missing. These findings indicate the high quality of the P. vachelli genome assembly (Table 6).
Gene annotation validation
To evaluate the integrity of the annotated gene set, we conducted BUSCO analysis using conserved single-copy homologous genes from the actinopterygii_odb10 library. The results revealed that approximately 96.54% of the complete gene elements are present in the annotated gene set, indicating a high level of completeness in the conserved gene predictions. Specifically, 95.08% of the genes were complete and single-copy BUSCOs, with only 0.47% genes fragmented and 2.99% genes missing from the assembly (Table 7). These findings highlight the exceptional integrity and conservation of gene content in the dace genome assembly, leading to highly confident prediction outcomes.
Code availability
No special codes or scripts were used in this work, and Data processing was carried out based on the protocols and manuals of the corresponding bioinformatics software.
Change history
20 March 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41597-025-04795-2
References
Liu, Y. et al. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 111, 1258–1265 (2019).
Zhang, G. et al. A high-density SNP-based genetic map and several economic traits-related loci in Pelteobagrus vachelli. Bmc Genomics 21, 1–17 (2020).
Zhang, G. et al. Effect of water temperature on sex ratio and growth rate of juvenile Pelteobagrus fulvidraco, P. vachelli and hybrids [P. fulvidraco (♀)× P. vachelli (♂)]. Aquaculture Reports 3, 115–119 (2016).
Zheng, K. et al. Effects of dietary lipid levels on growth, survival and lipid metabolism during early ontogeny of Pelteobagrus vachelli larvae. Aquaculture 299, 121–127 (2010).
Zheng, X. et al. Effects of hypoxic stress and recovery on oxidative stress, apoptosis, and intestinal microorganisms in Pelteobagrus vachelli. Aquaculture 543, 736945 (2021).
Zhang, G. et al. The effects of water temperature and stocking density on survival, feeding and growth of the juveniles of the hybrid yellow catfish from Pelteobagrus fulvidraco (♀)× Pelteobagrus vachelli (♂). Aquac Res 47, 2844–2850 (2016).
Hu, W.-H., Dan, C., Guo, W.-J. & Mei, J. The morphology and gonad development of Pelteobagrus fulvidraco and its interspecific hybrid” huangyou no. 1” with Pelteobaggrus vachelli. (2019).
Pei, X. et al. Effects of acute hypoxia and reoxygenation on oxygen sensors, respiratory metabolism, oxidative stress, and apoptosis in hybrid yellow catfish “Huangyou-1. Fish Physiology and Biochemistry 47, 1429–1448 (2021).
Mei, J. & Gui, J.-F. Genetic basis and biotechnological manipulation of sexual dimorphism and sex determination in fish. Science China Life Sciences 58, 124–136 (2015).
Mei, J. & Gui, J. F. Sexual size dimorphism, sex determination, and sex control in yellow catfish. Sex control in aquaculture, 495-507 (2018).
Gong, G. et al. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. GigaScience 7, giy120 (2018).
He, W.-P. et al. Chromosome-level genome assembly of the Chinese longsnout catfish Leiocassis longirostris. Zoological Research 42, 417 (2021).
Zhu, C. et al. Insights into chromosomal evolution and sex determination of Pseudobagrus ussuriensis (Bagridae, Siluriformes) based on a chromosome-level genome. DNA Res 29, dsac028 (2022).
Shao, F. et al. Chromosome-level genome assembly of the Asian red-tail catfish (Hemibagrus wyckioides). Frontiers in Genetics 12, 747684 (2021).
Zhou, Y.-L. et al. Barbel regeneration and function divergence in red-tail catfish (Hemibagrus wyckioides) based on the chromosome-level genomes and comparative transcriptomes. International Journal of Biological Macromolecules 232, 123374 (2023).
Ye, H. et al. Chromosome-level genome assembly of the largefin longbarbel catfish (Hemibagrus macropterus). Frontiers in Genetics 14, 1297119 (2023).
Yang, Y. et al. Gap-free chromosome-level genomes of male and female spotted longbarbel catfish Hemibagrus guttatus. Scientific Data 11, 572 (2024).
Gong, G. et al. Origin and chromatin remodeling of young X/Y sex chromosomes in catfish with sexual plasticity. National Science Review 10, nwac239 (2023).
Xiong, Y. et al. Biotechnological manipulation of the transition from genetic to temperature-dependent sex determination to obtain high quality neomale in aquaculture. Aquaculture 560, 738471 (2022).
Xiong, Y. et al. Sexually Dimorphic Gene Expression in X and Y Sperms Instructs Sexual Dimorphism of Embryonic Genome Activation in Yellow Catfish (Pelteobagrus fulvidraco). Biology 11, 1818 (2022).
Huang, L. et al. Profiling genetic breeding progress in bagrid catfishes. Fishes 8, 426 (2023).
Liu, H. et al. Genetic manipulation of sex ratio for the large-scale breeding of YY super-male and XY all-male yellow catfish (Pelteobagrus fulvidraco (Richardson)). Mar Biotechnol 15, 321–328 (2013).
Wang, D., Mao, H. L., Chen, H. X., Liu, H. Q. & Gui, J. F. Isolation of Y‐and X‐linked SCAR markers in yellow catfish and application in the production of all‐male populations. Anim Genet 40, 978–981 (2009).
Yu, Y. et al. High temperature-induced masculinization in yellow catfish Tachysurus fulvidraco: A potential approach for environmental-friendly mono-sex production. Aquaculture 534, 736263 (2021).
Gong, G. et al. A chromosome-level genome assembly of the darkbarbel catfish Pelteobagrus vachelli. Scientific Data 10, 598 (2023).
Li, J. et al. A high-quality chromosome-level genome assembly of Pelteobagrus vachelli provides insights into its environmental adaptation and population history. Frontiers in Genetics 13, 1050192 (2022).
Chen, S. Ultrafast one‐pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2, e107 (2023).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics 25, 1754–1760 (2009).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Wang, X. & Wang, L. GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Frontiers in plant science 7, 215951 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res 38, e199–e199 (2010).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 25, 4–10 (2009).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Gene prediction: Methods and protocols 161-177 (2019).
Chen, X. et al. High-quality genome assembly of channel catfish, Ictalurus punctatus. Gigascience 5, s13742–13016 (2016).
Zheng, S. et al. Chromosome‐level assembly of southern catfish (silurus meridionalis) provides insights into visual adaptation to nocturnal and benthic lifestyles. Mol Ecol Resour 21, 1575–1592 (2021).
Gao, Z. et al. A chromosome-level genome assembly of the striped catfish (Pangasianodon hypophthalmus). Genomics 113, 3349–3356 (2021).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22 (2008).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78–e78 (2015).
Bairoch, A. et al. The universal protein resource (UniProt). Nucleic Acids Res 33, D154–D159 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439 (2006).
NCBI https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1000294/ (2024).
NCBI Genbank https://identifiers.org/ncbi/insdc.gca:GCA_033026395.1 (2024).
Liu, H. et al. A chromosome-level genome assembly of the male darkbarbel catfish (Pelteobagrus vachelli). https://doi.org/10.6084/m9.figshare.26968447 (2024).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Tang, H. et al. JCVI: A versatile toolkit for comparative genomics analysis. iMeta, e211 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This work is supported by China Agriculture Research System (CARS-46); Central Public-interest Scientific Institution Basal Research Fund, CAFS (2023TD37); China-ASEAN Maritime Cooperation Fund (CAMC-2018F); Guangdong Province Rural Revitalization Strategy Special Fund (2023-SJS-00-001); National Freshwater Genetic Resource Center (FGRC18537); Guangdong Rural Revitalization Strategy Special Provincial Organization and Implementation Project Funds (2022-SBH-00-001); Natural Science Foundation of Jiangsu Province (BK20211367).
Author information
Authors and Affiliations
Contributions
H.L. and M.O. designed the study and led the research, J.Z., T.C. and X.Z. contribute to the materials of this study, H.L. and J.Z. analysed and uploaded the data, H.L. and L.F. contribute to the genome assembly and annotation, H.L., J.Z. and T.C. wrote the manuscript. All authors read and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, H., Zhang, J., Cui, T. et al. A chromosome-level genome assembly of the male darkbarbel catfish (Pelteobagrus vachelli) using PacBio HiFi and Hi-C data. Sci Data 12, 351 (2025). https://doi.org/10.1038/s41597-025-04662-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04662-0
