
Abstract
This study investigates the metabolic changes induced by endurance exercise, specifically race walking, in a cohort of 19 athletes. Blood samples were collected at four time points: pre-exercise (REST), immediately post-exercise (STAT), 3 hours into recovery (REC3), and 22 hours post-exercise (REC22). A total of 859 metabolites were identified through the untargeted method, and 465 metabolites and 411 lipids were identified through the targeted methods. Rigorous quality control measures were implemented throughout the study to ensure data reliability. The comprehensive dataset, which is publicly available on the Metabolomics Workbench website, offers valuable insights into the systemic metabolic shifts triggered by endurance exercise. This resource may prove instrumental in uncovering biomarkers associated with athletic performance, providing a foundation for future research in exercise physiology and metabolic health.
Similar content being viewed by others

Background & Summary
Race walking is an endurance exercise featured in the World Athletics Championships and the Olympic Games1. Endurance sports are widely participated in worldwide and have been proven highly beneficial for health2. Physical activity augments musculoskeletal function3 and reduces the risk of obesity4, diabetes5, chronic kidney disease6 and cancer7. The study of metabolic states during endurance sports may help to understand how the body efficiently utilizes energy during prolonged periods, which is essential for athletes and individuals engaging in sustained physical activity. Also, insights into the endurance metabolism can inform strategies to optimize performance and recovery, enhancing athletic achievements. Moreover, research in this field contributes to our understanding of overall metabolic health, potentially offering insights into combating metabolic disorders and promoting general well-being.
A large body of literature has demonstrated the potential of metabolomics in exercise, suggesting that metabolomic markers can help unravel the complex biochemical pathways that link exercise to disease. Lewis et al. demonstrated that the concentrations of niacinamide, glucose-6-phosphate, pantothenate, and succinate had increased levels in plasma after exercise and provided insights into a profile of exercise performance and susceptibility to cardiovascular disease, in addition to highlighting molecular pathways that might modulate the beneficial effects of exercise8. Andrew et al. integrated metabolomics and transcriptomics data and found that changes in glucose utilization induced by regular exercise appear to be important for maintaining mitochondrial health and deriving maximum cardiac benefits from physical activity9. These studies have led to a broader collection of metabolomics data to validate the effectiveness of exercise.
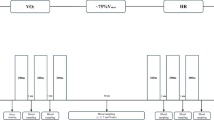
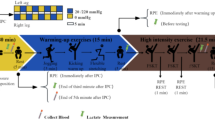
Here, we present a new dataset related to 19 individuals from Dalian City race walking team, for which metabolomic profiling was performed on plasma samples collected (Fig. 1).The metabolite quantification included six profiling modes:(1) one Hydrophilic Interaction Chromatography (HILIC) ultra-high performance liquid chromatography (UPLC)-MS/MS using both positive and negative ion mode electrospray ionization (ESI); (2) one independent reversed phase (RP)/ UPLC -tandem mass spectrometry (MS/MS) method using negative ion mode electrospray ionization (ESI); (3) one RP chemical derivatization method, using negative ion mode electrospray ionization (ESI); (4–6) three independent RP/UPLC-MS/MS with both positive negative ion mode ESI that allow the detection of more than 1800 compounds across 42 molecular classes.

Overview of the experimental process.
Here we make these data, together with the corresponding rich clinical annotations, publicly available to the scientific community.
Methods
Sample collection: (study design)
Sera were collected from 20 male student-athletes with the informed consent of all participant, and the study was approved by the ethics committee of Shenyang Sport University (No.2022-6-30-01). Samples were collected at four different time points. The first point was fasting samples collected before the race walk at 8:20 am (named REST). The second point was a sample collected immediately after the race walk exercise at 10:30 am (named STAT). The third point was collected during the 3-hour recovery period after the exercise at 13:30 pm (named REC3). The fourth point was fasting samples collected at 22 h post-exercise on day 2 at 8:20 am (named REC22). Blood was collected in plastic Vacutainer blood tubes, allowed to clot for 40 minutes, and then centrifuged at 13000 g for 10 minutes. The sera were frozen on dry ice immediately after collection and then stored at −80 °C until analysis. One athlete chose to exit the study (No. 80) and so their readings are omitted and the data on heart rates at each time point are shown in Table 1.
Sample pretreatment of metabolites
The aliquots were thawed at 4 °C for 60 minutes and then gently pipetted with a pipetting gun to allow uniform distribution of the sample. Then, 120 μL of serum was transferred to a 2 mL EP tube (Aixgen USA), and 480 μL of methanol was added. The samples were vortexed for 5 min then were allowed to stand for 10 min at room temperature and centrifuged at 13000 g at 4 °C (Thermo Scientific, USA) for 10 minutes. The upper extracts were transferred to a 1.5 mL tube and lyophilized for 3 hours in a vacuum freeze dryer (Labconco Corporation, USA) and then stored at 4 °C until injection. Prior to analysis, the extracts were redissolved in 500 μL acetonitrile-water (1:3, v/v). Quality control (QC) samples were prepared by mixing equal amounts of serum samples.
Sample pretreatment for lipidomics
Firstly, 2 mL lipid internal standards (LIS) were prepared which consist of 100 ng/mL phosphatidylethanolamine (PE) (15:0-18:1-d7; 1.1 μg/mL triglyceride (TG) 15:0-18:1-d7-15:0; C16 218 ng/mL ceramide-d7 (d18:1-d7/16:0); 1 μg/mL stearic acid d35; 4 μg/mL lysophosphatidylinositol (LPI) 17:0-d5 (From Avanti Polar Lipids, Inc. Alabaster, AL, USA). 20 μL serum sample was transferred to a 2 mL tube, then 120 μL LIS and 200 μL methanol were added. The mixture was blown gently with a pipette. After vortexing for 5 minutes, 360 μL methyl tert-butyl ether (MTBE) and 100 μL ultrapure water were added and then vortexed for 3 minutes. The mixture was allowed to stand for 3 minutes at 4 °C, and then centrifuged at 4 °C, 5300 rpm × 10 min. 300 μL of the upper layer was transferred to a new tube and freeze-dried. The samples were stored at 4 °C until analysis. Lipid samples were redissolved in acetonitrile-isopropanol (1:1, v/v) solution before injection.
Sample pretreatment for targeted metabolomics
The targeted metabolomics detection method we used was based on the deep targeted metabolomics analysis platform established by our laboratory10. For targeted metabolomics, the pretreatment steps were as follows:
Sample pretreatment for Amino compounds (M1)
Firstly, the stock solution of mixed internal standard was diluted to 10 different concentrations for quantitative curves. Transfer 50 μL sample to 750 μL 96-well plate, add 10 μL HILIC-POS IS and 190 μL methanol for vortexing (1500 rpm × 3 min). The supernatant was then centrifuged (5300 rpm × 20 min) for LC-MS/MS analysis.
Sample pretreatment for bile acids and fatty acids (M2)
50 μL of samples were transferred to EP tubes, and 20 μL of IS was added. After mixing with 180 μL methanol-acetonitrile (1:1, v/v), the supernatant was vortexed (1300 rpm × 5 min), and then centrifuged (12000 g × 20 min). 180 μL of the supernatant was used for LC-MS/MS testing.
Sample pretreatment for carbohydrate pretreatment (M3)
20 μL of samples were transferred to a 1.5 mL EP tube, and then 80 μL methanol-acetonitrile (1:1, v/v) was added and vortexed (1300 rpm × 5 min). After centrifugation, 40 μL (12000 rpm × 20 min) of the supernatant was taken into another 1.5 mL EP tube in the middle. 5 μL GlycoM-IS and 20 μL derivatization reagent (200 mM 3-NPH) and 20 μL catalyst (200 mM EDC,6% pyridine) were added to the EP tube and vortexed (30 °C, 1000 rpm × 30 min). Subsequently, the tubes were dried in a vacuum freeze dryer (Labconco, USA) for 30 min. The dried EP tubes were redissolved in 800 μL 50% methanolic water and vortexed (1500 rpm × 5 min). After centrifugation (12000 rpm × 20 min), the supernatant was used for LC-MS/MS testing.
Sample pretreatment for gut microbiota metabolites (M4)
50 μL of samples were transferred to 750 μL 96-well plate, and 10 μL gut microbiota IS and 190 μL methanol-acetonitrile = 1:1(1:1, v/v) were added and vortexed (1500 rpm × 3 min). After centrifugation (5300 rpm × 20 min), 120 μL of supernatant was diluted with 120 μL ultrapure water. After vortexing (1500 rpm × 3 min) and centrifugation (5300 rpm × 20 min), 180 μL supernatant (5300 rpm × 20 min) was injected.
Sample pretreatment for phospholipids and ACCA (M5)
50 μL samples were transferred to 750 μL Waters 96-well plate, and 50 μL internal standard mixture AcCa + Lyso-P IS and 150 μL methanol were added for vortexing (1300 rpm × 3 min). After vortexing and centrifugation (5300 rmp × 20 min), 180 μL of the supernatant was used for LC-MS/MS testing.
Instrument parameters for untargeted metabolomics
Untargeted metabolomics was performed on an Ultimate 3000 ultra-performance liquid chromatography coupled to a Q-Exactive modular quadrupole-orbital ion trap mass spectrometer (Thermo Scientific, United States).
Extracts were separated reversed-phase chromatography with an injection volume of 5 μL using an AcquityTM HSS C18 column (Waters Co., USA, 2.1 × 100 mm) for the first class of metabolites. Mobile phases were A (0.1% formic acid/water) and B (0.1% formic acid/acetonitrile), and the separation gradient rose from an initial 2% B to 98% within 10 min. An AcquityTM BEH C18 column (Waters Co., USA, 1.7 μm, 2.1 × 100 mm) was used for the other class of metabolites. Mobile phase A is water and B is water /acetonitrile 5:95 v/v, both containing ammonium bicarbonate (400 mg/L). The initial 2% B rose to 100% within 10 min. The following 5 min was used to wash and equilibrate the column. The flow rate was 0.4 mL/min, and column temperature was 50 °C.
Instrument parameters for mass detection were set to the following: The sheath gas flow rate was 45 arb, the auxiliary gas flow rate was 10 arb, the ionization chamber heating temperature was 355 °C, the capillary temperature was 320 °C, and the S-Lens RF level was 55%. Metabolites were analyzed in full scan mode with a full scan resolution of 70000 FWHM, a mass-to-charge ratio scan range set between 70 and 1000 m/z. For secondary mass spectrometry MS/MS qualitative data acquisition 17500 FWHM resolution was used. Tip-triggering, dynamic repulsion, and isotope repulsion modes were enabled, and the precursor separation window was set to 1.0 Da. The data obtained with all the above parameters were acquired through collision-induced dissociation of metabolites using ultrapure nitrogen as the cleavage gas.
Instrument parameters for targeted metabolomics
For targeted metabolomics assays, quantitation was performed using a Shimadzu LC-20ADXR (Shimadzu, Japan) tandem Sciex 5500 + triple quadrupole mass spectrometer (Sciex, Singapore). The metabolites were ionized by TurboVTM heated electrospray ionization source, and then detected by scheduled multiple reaction monitoring mode (MRM).
For the separation of amino acids, an Acquity UPLC BEH Amide column (1.7 μm, 2.1 × 100 mm, Waters Inc.) was used with 2 μL injection, 90% aqueous acetonitrile as mobile phase A and 50% aqueous acetonitrile as mobile phase B, and ammonium formate and formic acid were added as buffer salts to improve the separation. The chromatographic gradient was from 0% B to 70% B, the elution time was 15 min, the flow rate was maintained at 0.3 mL/min.
For bile acid and fatty acid separation, a Hypersil GOLD column (1.9 μm, 2.1 × 100 mm, Thermo Fisher Scientific Inc.) was used, and 2 μL of sample was injected. 2 mM ammonium acetate was added to water as mobile phase A, acetonitrile as mobile phase B, and the gradient was 0.5–12 min 0–17%B, respectively. 12–15.5 min, 17–30% B; 15.5–16.5 min, 55% B; 16.5–24.5 min, 55–95% B; 24.5–27.0 min, 95% B, flow rate was maintained at 0.4 mL/min, injection volume was 3 μL.
For the separation of carbohydrates, an ACQUITY UPLC HSS C18 column (1.8 μm, 2.1 × 100 mm, Waters Inc.) was used, with 2 μL of sample injected, 0.1% formic acid-water as mobile phase A, acetonitrile as mobile phase B, and a flow rate of 0.3 mL/min. The gradient increased from 0% to 95% in 23 min. The injection volume was 2 μL.
For the targeted separation of gut microbiota metabolites, Excel PFP C18 column (2 μm, 2.1 × 100 mm, ACE Inc.) was used, 2 μL of sample was injected, 0.1% formic acid-water was used as mobile phase A, acetonitrile was used as mobile phase B, and the flow rate was 0.3 mL/min. The gradient increased from 5% to 95% in 9.5 min. The column temperature was 40 °C, and the injection volume was 5 μL.
For the targeted separation of acylcarnitines and lysophospholipids, the metabolites were separated on an Excel PFP C18 column (2 μm, 2.1 × 100 mm, ACE Inc.) and 0.1% formic acid in water as mobile phase A, and 0.1% formic acid in acetonitrile as mobile phase B. The chromatographic gradient ramped from 2% strong elution to 98% in 12 min. Meanwhile, the flow-rate was set at 0.4 mL/min 5 μL aliquots of metabolites were injected.
The main mass parameters were optimized as follows: negative IonSpray voltage −4.5 kV; Curtain gas: 35 psi; Ion gas 1 and 2: 50 psi; heater temperature: 550°C; positive IonSpray voltage 5.5 kV; Curtain gas: 35 psi; Ion gas 1 and 2: 50 psi; heater temperature: 550 °C.
Instrument parameters for lipidomics
Lipid analysis was performed similarly on a 4500 triple quadrupole mass spectrometer (Sciex, Singapore). The injection volume of lipids was 2 μL and the separation was performed on an ACQUITY HSS C18 column (1.8 μm, 2.1 × 100 mm, Waters Inc.) at the column temperature of 40 °C. Mobile phase A consisted of acetonitrile/water (60:40 v/v, 5 mM ammonium formate) and mobile phase B consisted of isopropanol/acetonitrile (90:10 v/v, 5 mM ammonium formate). The chromatographic separation flow rate was 0.2 mL/min, and the mobile phase gradient rose from an initial 20% B to 95% within 17.5 min.
Ionization parameters: Turbo V electrospray ion source, positive ionization voltage 5.5 kV, ion source temperature 550 °C, curtain gas 35 psi, GS1 50 psi, GS2 50 psi; The ionization voltage of the negative ion was −4.5 kV, the ion source temperature was 550 °C, and the curtain gas was 35 psi, GS1 50 psi, and GS2 50 psi.
Data analysis
Metabolic peak alignment and extraction were performed using Compound Discoverer software (Thermo Scientific, USA). The structural annotation was followed the instructions of MSI11, using our inhome database (iPhenomeTM SMOL high-resolution MS/MS spectrum library12), NIST 17 Tandem MS/MS library13 (US National Institute of Standards and Technology), mzCloud library14 (Thermo Scientific, USA), Human Metabolome Database15 (HMDB), and Kyoto Encyclopedia of Genes and Genomes16 (KEGG). The peak areas of annotated metabolites were extracted by TraceFinder software (Thermo Scientific, San Jose, USA). Mass accuracy was controlled within ± 5 ppm for metabolite identification or structure annotation, and the chemical formula was confirmed by introducing isotope information within 10 ppm for at least one isotope and a fitting fraction of 70% of the relative isotope abundance map on the basis of the exact mass. In addition, the structural annotation of metabolites was rigorously confirmed using retention time information as well as high-resolution MS/MS spectrum similarity.
Targeted metabolites were collected by selective reaction detection mode SRM and quantified by isotope labeled internal standard method. We compared the primary and secondary mass spectrometry data of the targeted metabolites with the standards. We calculated quantitative data of each sample by combining the standard curve and the area under the curve (AUC) values. Finally, internal standards were used for calibration. The above analyses were conducted with Analyst and OS-MQ software version B.06.00 (SCIEX, Singapore).
Lipid Search (Thermo Scientific™) software was used to collect the data of non-targeted lipidomics by selective reaction detection mode SRM, and isotope labeled internal standard method was used for quantification. The data were processed, including peak extraction and identification of lipid compounds, etc. TraceFinder software (Thermo Scientific) was used to extract the area under the curve (AUC) as the relative quantitative information of metabolites and lipids, and strict manual inspection and one-by-one inspection were performed to eliminate false positives mainly based on peak shaking, adduct ion behavior, fragmentation pattern, and chromatographic behavior. All peak area data for labeled metabolites were exported to Excel software for trimming.
Metabolites with missing values of more than 50% of the sample number were excluded in the statistical analysis when metabolomic data were analyzed. The remaining missing values were estimated and populated using the K-nearest neighbour algorithm17. To make the metabolomics data reproducible, the relative standard deviation (RSD) of peaks greater than 30% in QC samples was filtered out to filter molecules with an RSD higher than 30%. Principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) were performed using Metaboanalyst v6.0 software18 and analysis of variance (ANOVA) was applied to calculate statistical significance (p-values). The following criteria were used to identify differential metabolites: variable importance in projection” (VIP) (VIP) >1 and p-values < 0.05, log2 (fold change) ≥ 1 or ≤−1.
Data Records
The integrated metabolite ion counts, the preprocessed metabolomics data, the metabolite annotations are available for download on Metabolomics Workbench, https://doi.org/10.21228/M8C80219. The data is available as a zip file (walkersMZ.zip, targeted-walkers.zip, and Targeted Metabolomics Raw Data.zip).
Technical Validation
QC (quality control) samples were used for technical validation. Unsupervised principal component analysis (PCA) was applied to identify the main source of variation within the data20 (Fig. 2a), where it can be observed that QCs tend to cluster together. More than 99% of the analytes in the untargeted metabolomics and targeted metabolomics data exhibited a relative coefficient of variation RSD (%) of less than 25% in the QC samples (Fig. 2B). The stability of the first principal component of QCs over time is demonstrated in Fig. 2C. In addition, Spearman correlation analysis between the first and last QC data had a high correlation (Fig. 2D).

Technical validation data for QC samples in methods. (a) Principal component analysis (PCA) of cohort samples. Including untargeted metabolomics; targeted metabolomics and lipidomics. (b) Distribution graph of relative variation coefficient for the analytes in QCs. Including untargeted metabolomics; targeted metabolomics and lipidomics. (c) The stability of the first principal component of QCs over time. Including untargeted metabolomics; targeted metabolomics and lipidomics. (d) Spearman correlation analysis between the first and last QC data.
Code availability
No custom code was used to generate or process the data described in the manuscript.
References
Burke, L. M., Jeukendrup, A. E., Jones, A. M. & Mooses, M. Contemporary Nutrition Strategies to Optimize Performance in Distance Runners and Race Walkers. Int J Sport Nutr Exerc Metab 29, 117–129, https://doi.org/10.1123/ijsnem.2019-0004 (2019).
Kelly, R. S., Kelly, M. P. & Kelly, P. Metabolomics, physical activity, exercise and health: A review of the current evidence. Biochim Biophys Acta Mol Basis Dis 1866, 165936, https://doi.org/10.1016/j.bbadis.2020.165936 (2020).
Egan, B. & Zierath, J. R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184, https://doi.org/10.1016/j.cmet.2012.12.012 (2013).
Mielke, G. I., Bailey, T. G., Burton, N. W. & Brown, W. J. Participation in sports/recreational activities and incidence of hypertension, diabetes, and obesity in adults. Scand J Med Sci Sports 30, 2390–2398, https://doi.org/10.1111/sms.13795 (2020).
Khasanova, A. & Henagan, T. M. Exercise Is Medicine: How Do We Implement It? Nutrients 15, https://doi.org/10.3390/nu15143164 (2023).
Baiao, V. M. et al. Effects of Exercise on Inflammatory Markers in Individuals with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Metabolites 13, https://doi.org/10.3390/metabo13070795 (2023).
Hardt, L. M. et al. Are Guideline Recommendations on Supportive Nutrition and Exercise Therapy for Cancer Patients Implemented in Clinical Routine? A National Survey with Real-Life Data. Nutrients 15, https://doi.org/10.3390/nu15143172 (2023).
Lewis, G. D. et al. Metabolic signatures of exercise in human plasma. Sci Transl Med 2, 33ra37, https://doi.org/10.1126/scitranslmed.3001006 (2010).
Gibb, A. A. et al. Exercise-Induced Changes in Glucose Metabolism Promote Physiological Cardiac Growth. Circulation 136, 2144–2157, https://doi.org/10.1161/CIRCULATIONAHA.117.028274 (2017).
Ma, S. R. & Yin, P. Y. Dataset of the comprehensive description and methodological validation for an in-depth targeted metabolomics platform developed by Yin Lab. Science Data Bank, https://doi.org/10.57760/sciencedb.13920 (2024).
Members, M. S. I. B. et al. The metabolomics standards initiative. Nat Biotechnol 25, 846–848, https://doi.org/10.1038/nbt0807-846b (2007).
Ye, G. et al. Comprehensive metabolic responses of HepG2 cells to fine particulate matter exposure: Insights from an untargeted metabolomics. Sci Total Environ 691, 874–884, https://doi.org/10.1016/j.scitotenv.2019.07.192 (2019).
Rahme, M. et al. Limitations of platform assays to measure serum 25OHD level impact on guidelines and practice decision making. Metabolism 89, 1–7, https://doi.org/10.1016/j.metabol.2018.09.003 (2018).
Yi, Z. & Zhu, Z. J. Overview of Tandem Mass Spectral and Metabolite Databases for Metabolite Identification in Metabolomics. Methods Mol Biol 2104, 139–148, https://doi.org/10.1007/978-1-0716-0239-3_8 (2020).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res 46, D608–D617, https://doi.org/10.1093/nar/gkx1089 (2018).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–462, https://doi.org/10.1093/nar/gkv1070 (2016).
Do, K. T. et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics 14, 128, https://doi.org/10.1007/s11306-018-1420-2 (2018).
Pang, Z. et al. MetaboAnalyst 6.0: towards a unified platform for metabolomics data processing, analysis and interpretation. Nucleic Acids Res 52, W398–W406, https://doi.org/10.1093/nar/gkae253 (2024).
Zhang, Y. et al. An integrated LC-MS analysis of the biometric characteristics of different time cohorts of race walkers. Metabolomics Workbench. https://doi.org/10.21228/M8C802 (2024).
Feng, L. et al. Effects of heat stress on 16S rDNA, metagenome and metabolome in Holstein cows at different growth stages. Sci Data 9, 644, https://doi.org/10.1038/s41597-022-01777-6 (2022).
Acknowledgements
We thank all the participants from Dalian Sports Development Center. The work was funded by 2021 Scientific Research Funding Project of Liaoning Provincial Department of Education (No: LJKR0475).
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.Wu, J.Zhou, P.Yin; methodology, Z.Wu.; analysis, J.Lai, Y.Zhang, S.Ma; investigation, Y.He, Y.Zhang; resources, Y.He, J.Zhou; writing—original draft preparation, Y.Zhang.; writing—review and editing, S.Ma, P.Yin.; supervision, P.Yin. Z.Wu; project administration, Z.Wu, J.Zhou; All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Z.Wu and P.Yin are cofounders of IPhenome Biotechnology (Yunpukang) Inc, Dalian. Other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, Y., Zhang, Y., Lai, J. et al. Plasma metabolomics dataset of race-walking athletes illuminating systemic metabolic reaction of exercise. Sci Data 12, 448 (2025). https://doi.org/10.1038/s41597-025-04751-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04751-0
