
Abstract
P. serrula is widely distributed in Yunnan, Xizang, and Sichuan, and usually grows at high altitudes between 2,600 and 3,900 meters above sea level. In this study, we obtained a high-quality chromosome-level assembly genome of P. serrula using Illumina sequencing, Oxford Nanopore ultra-long sequencing, and Hi-C technology. The genome was 284.5 Mb in length, with a scaffold N50 of 32.4 Mb and 91.9% of the assembly anchored onto 8 pseudochromosomes. BUSCO completeness value of 98.5% demonstrated a high completed genome, and a total of 35,151 protein-coding genes and 47,340 transcripts were annotated. Overall, this genome delivers valuable genetic resources for further phylogenomic studies and provides insights into the genetic architectures underlying high-altitude adaptation.
Similar content being viewed by others

Background & Summary
With approximately 3,000 species and 88–100 genera, Rosaceae is one of the most diverse angiosperm-family genera worldwide1,2. The Rosaceae have a wide variety of fruit types2,3, including Strawberry, Raspberry, Apple, Pear, Peach, Apricot, Plum, and Cherry. Due to the important economic value of these fruits, their production has increased rapidly in the past decade, for example, the production of plums has also increased from 5.52 million tons in 2010 to 6.63 million tons in 2021. In the meantime, many researchers have reported the evolutionary history of this family4,5. However, due to the wide variety of species in the Rosaceae family and the frequent occurrence of hybridization events, the evolutionary history of this family is still unclear.
Prunus is a shrub or tree of Rosaceae mainly distributed in the north temperate zone, with about 30 species6. In addition to important economic value, some species of Prunus also have high ornamental value, such as Prunus mira, Prunus persica, Prunus mume, and Prunus yedoensis. To date, many Prunus genomes have been released: P. persica, P. mira, Prunus dulcis, Prunus ferganensis, Prunus davidiana, Prunus mume ‘Xizang’, Prunus armeniaca ‘Xizang’, Prunus salicina, Prunus humilis, Prunus domestica, P. yedoensis, and Prunus avium7,8,9,10,11,12,13,14,15,16,17,18. However, the Xizang cherry genome has not been reported yet.
The Tibetan Plateau has an average elevation of more than 4,000 m, and with that comes an extremely harsh environment, such as high UV-B radiation, low temperatures, low oxygen content, and low barometric. In addition, the Tibetan Plateau contains many wild Prunus resources; thus, these wild Prunus resources were not only selected by humans but also by the environment. Up to date, many studies have been done on the mechanism of high-altitude adaptation. For example, genomic selective scavenging analysis of two subgroups at high and low altitudes of P. mira demonstrated that the selected genes were functionally enriched in response to UV-B radiation16; Comparative population analysis indicated that a CBF gene might be the key factor in the adaptation of P. mira to low temperatures at high altitudes19. Even so, the effects of a high-altitude environment on genomic variation are poorly understood. Moreover, given the abundance of many wild cherry resources on the Qinghai-Tibet Plateau, how these resources adapt to high altitudes has not been reported.
P. avium is a fruit crop that grows agronomically and economically in the Rosaceae family, and this species usually grows in temperate climatic areas to provide the chilling requirement necessary for flower induction20,21. P. avium originated probably between the Black Sea and the Caspian and then spread to European temperate regions22. To date, more than thirty cherry species have been identified, resulting in diverse phenotypic variations in fruit, size, color, and other important agronomic traits23,24. In addition, previous genetic analysis studies demonstrated that a narrow genetic bottleneck occurred in modern cultivars23,25. However, little is known about the phenotype and genetic variation of Xizang cherry resources.
In this study, we assembled a high-quality chromosome-level P. serrula genome using Oxford Nanopore ultra-long reads and chromosome conformation capture sequencing (Hi-C). In conclusion, this genome provides valuable genetic resources for underlying the high-altitude adaptation of the Prunus fruit tree.
Methods
Materials collection and sequencing
Fresh young leaves used for genome sequencing were collected from the P. serrula plant grown in the wild environment of Xizang, China. The total genomic DNA for each of the accessions was extracted from leaves using the CTAB method26. The DNA-seq was performed on the Illumina NovaSeq 6000 platform.
Transcriptome sequencing was performed on the mixed samples of the three tissues (fruit, leaves, branches) for genome annotation. Total RNA was extracted using the RNAprep Pure Plant Kit (DP441, TIANGEN Biotech). RNA-seq was conducted on the Illumina NovaSeq 6000 platform, and 150-bp paired-end reads were generated. Hi-C libraries were controlled for quality and sequenced on an Illumina Novaseq platform with 150 bp paired-end reads.
De novo assembly and annotation of three Prunus genomes
The RepeatModeler software27 was used to build a mixed de novo TE library based on the genomes of diploid Xizang cherry, tetraploid Xizang cherry, and hexaploidy Xizang plum. This TE library and the Repbase database (https://www.girinst.org/repbase/) were used to annotate repeat sequences using RepeatMasker28.
Gene models were annotated based on ab initio gene predictions, protein homology searches, and RNA-seq reads based transcript assemblies. For ab initio gene predictions, AUGUSTUS29, GlimmerHMM30, and SNAP31 were employed using default parameters. The protein databases were constructed by integrating the amino acid sequences from the Rosaceae databases. Homology searching was then conducted using GenomeThreader32. In addition, RNA-seq reads were generated from a mixture of tissues. The Trinity software33 was used to perform genome-guided and de novo transcript assembly. The PASA software34 was used to update the protein-coding gene annotations. All of the gene structures predicted were combined using the EVM software35.
We assembled a high-quality genome with an N50 value of 9.5 Mb and the longest contig size of 19.5 Mb. The error correction of contigs was performed using Racon36 and was iterated three times based on Nanopore reads, followed by two rounds of polishing using NextPolish37 with Illumina short reads. With the Hi-C library, the error-corrected contigs were anchored to eight superscaffolds using the tools 3D-DNA38 and juicer39. The analysis of Benchmarking Universal Single-Copy Orthologs (BUSCO) revealed40 a completeness score of 98.5% (Table 1).
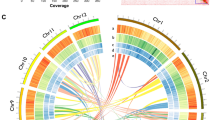
We annotated 35,151 protein-coding genes and 47,340 transcripts by combining ab initio prediction, RNA-Seq read mapping, and homologous protein alignments. To show the characteristics of the P. serrula genome, we identified Presence/Absence Variations (PAVs, which are genomic regions that are present in one genome but absent in another, representing structural variations that may contribute to phenotypic differences between species) between the P. serrula genome and the cultivated cherry genome41, and we also exhibited GC content, gene density, and TE density of the P. serrula genome (Fig. 1B).

De novo genome assembly and genome features of P. serrula (A) Phenotypic characteristics of P. serrula fruit. (B) Genome features of the Xiang cherry genome and the landscape of presence/absence variation (PAV) between the Xizang cherry genome and the cultivated cherry genome. The lines in the center of the circle indicate pairs of homologous genes on the different chromosomes of P. serrula.
Data Records
The whole-genome sequencing data (Table 2) were deposited to the NCBI Sequence Read Archive with accession number SRP45415942. The genome assembly data had been submitted to GenBank with accession number JBJZPD00000000043. The genome and genome annotation files of the P. serrula and two other Polyploid Xizang Prunus were also deposited to the Figshare database44,45.
Technical Validation
High completeness of genome assembly
99.4% of short reads and 99.5% of Nanopore ultra-long reads were remapped to the assembled P. serrula genome, we also conducted statistics on the BUSCO data for 44 Prunus genomes, these results demonstrated that our assembled genome was highly complete. Furthermore, the Hi-C contact map also suggested the result (Fig. 2). These evaluations reveal that genome assemblies are of high quality and suitable for use as reference genomes.

Hi-C contact matrix heatmap of the P. serrula genome.
Code availability
All software with their specific version used for data processing are clearly described in the methods section. If no specific variable or parameters are mentioned for a software, the default parameters were used.
References
Hummer, K.E., Janick, J. Rosaceae: Taxonomy, Economic Importance, Genomics. Plant Genetics and Genomics: Crops and Models. Springer (2009).
Morin, N. R., Brouillet, L. & Levin, G. A. Flora of North America North of Mexico. Rodriguésia 66, 973–981 (2015).
Potter, D. et al. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266, 5–43 (2007).
Xiang, Y. et al. Evolution of Rosaceae Fruit Types Based on Nuclear Phylogeny in the Context of Geological Times and Genome Duplication. Mol Biol Evol 34, 262–281 (2017).
Zhang, S. D. et al. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol 214, 1355–1367 (2017).
Zheng, T. et al. The chromosome-level genome provides insight into the molecular mechanism underlying the tortuous-branch phenotype of Prunus mume. New Phytol 235, 141–156 (2022).
Zhang, Q. et al. The genome of Prunus mume. Nat Commun 3, 1318 (2012).
Verde, I. et al. The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom 18, 225 (2017).
Baek, S. et al. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol 19, 127 (2018).
Zhang, Q. et al. The genetic architecture of floral traits in the woody plant Prunus mume. Nat Commun 9, 1702 (2018).
Jiang, F. et al. The apricot Prunus armeniaca L. genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Hortic Res 6, 128 (2019).
Alioto, T. et al. Transposons played a major role in the diversification between the closely related almond and peach genomes: results from the almond genome sequence. Plant J 101, 455–472 (2020).
Liu, C. et al. Chromosome-level draft genome of a diploid plum Prunus salicina. Gigascience 9 (2020).
Callahan, A. M. et al. Defining the ‘HoneySweet’ insertion event utilizing NextGen sequencing and a de novo genome assembly of plum Prunus domestica. Hortic Res 8, 8 (2021).
Tan, Q. et al. Chromosome-level genome assemblies of five Prunus species and genome-wide association studies for key agronomic traits in peach. Hortic Res 8, 213 (2021).
Wang, X. et al. Genomic basis of high-altitude adaptation in Tibetan Prunus fruit trees. Curr Biol 31, 3848–3860 e3848 (2021).
Fang, Z. Z. et al. The genome of low-chill Chinese plum “Sanyueli” Prunus salicina Lindl. provides insights into the regulation of the chilling requirement of flower buds. Mol Ecol Resour 22, 1919–1938 (2022).
Wang, Y. et al. The genome of Prunus humilis provides new insights to drought adaption and population diversity. DNA Res 29 (2022).
Cao, K. et al. Chromosome-level genome assemblies of four wild peach species provide insights into genome evolution and genetic basis of stress resistance. BMC Biol 20, 139 (2022).
Fernandez, I. M. A. et al. Genetic diversity and relatedness of sweet cherry prunus avium L. cultivars based on single nucleotide polymorphic markers. Front Plant Sci 3, 116 (2012).
Ganopoulos, I., Tsaballa, A., Xanthopoulou, A., Madesis, P. & Tsaftaris, A. Sweet Cherry Cultivar Identification by High-Resolution-Melting HRM. Analysis Using Gene-Based SNP Markers. Plant Mol Biol Rep 31, 763–768 (2012).
Blando, F. & Oomah, B. D. Sweet and sour cherries: Origin, distribution, nutritional composition and health benefits. Trends Food Sci Tech 86, 517–529 (2019).
Campoy, J. A. et al. Genetic diversity, linkage disequilibrium, population structure and construction of a core collection of Prunus avium L. landraces and bred cultivars. BMC Plant Biol 16, 49 (2016).
Zambounis, A. et al. Evidence of extensive positive selection acting on cherry Prunus avium L. resistance gene analogs RGAs. Aust. J. Crop Sci 10, 1324–1329 (2016).
Xanthopoulou, A. et al. Whole genome re-sequencing of sweet cherry Prunus avium L. yields insights into genomic diversity of a fruit species. Hortic Res 7, 60 (2020).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8, 4321–4325 (1980).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4 10 11–14 10 14 (2009).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinform 5, 59 (2004).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inform Software Tech 47, 965–978 (2005).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746 (2017).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Sys.t 3, 95–98 (2016).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654 (2021).
Wang, J. et al. Chromosome-scale genome assembly of sweet cherry (Prunus avium l.) cv. tieton obtained using long-read and hi-c sequencing. Hortic Res 7, 122 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP454159 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBJZPD000000000 (2024).
Zuo, H. Tibetan cherry genome. figshare https://doi.org/10.6084/m9.figshare.25036583.v1 (2024).
Zuo, H. Polyploid Tibetan Prunus genome. figshare https://doi.org/10.6084/m9.figshare.25037495.v1 (2024).
Acknowledgements
This work was supported by the Tibet Finance the Tibet Economic Forest Seedling Cultivation Project (202375), the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502).
Author information
Authors and Affiliations
Contributions
Xiuli Zeng, Wen-Biao Jiao, and Qiang Xu conceived and designed the project and the strategy. Xiuli Zeng, Yuanrong Li, Pingcuo Gesang, and Hong Ying collected the samples. Hao Zuo assembled the genome with the help of Lei Tan, Shengjun Liu and Yue Huang. Hao Zuo wrote the original paper, and Qiang Xu, Xia Wang, and Wen-Biao Jiao revised the original paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zuo, H., Liu, S., Tan, L. et al. Chromosome-level assembly of Prunus serrula Franch genome. Sci Data 12, 494 (2025). https://doi.org/10.1038/s41597-025-04810-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04810-6
