
Abstract
Eriophyoidea represents a highly diverse superfamily of herbivorous mites in the Acariformes, including over 5,000 named species that are distributed worldwide. However, the lack of chromosome-level genome prevents our understanding of the evolution in this group. Here, we report the first chromosome-level genome assembly of Setoptus koraiensis using Illumina, PacBio, and Hi-C sequencing technologies. The assembled genome has a size of 47 Mb with an N50 of 24.53 Mb, anchored into two chromosomes. The chromosome-level genome assembly had a BUSCO completeness of 89%. We identified 5,954 protein-coding genes, with 4,770 genes that could be functionally annotated. This genome provides resources to further understand the genetic and evolution of eriophyoid mites.
Similar content being viewed by others


Background & Summary
Eriophyoid mites (Acariformes, Eriophyoidea) are among the largest superfamilies in the Arachnida, comprising over 5,000 name species1,2 and exhibiting a worldwide distribution3. These tiny (~200 um in length, among the smallest arthropods), vermiform to fusiform mites have only two pairs of legs, and are strictly phytophagous, reflecting high hostplant specificity4,5; some of them can cause massive economic losses in agriculture and forestry6.
Despite the need to understand the ecology and evolution among eriophyoid mites, there are no chromosome-level assembled genomes for eriophyoid mites yet. A near chromosome genome assembly has been published for tomato russet mite Aculops lycopersici7, but the lack of high-quality chromosome-level genome resources has limited further comparative genomic analyses among eriophyoid mites.
In this study, we assembled a chromosome-level genome for the Setoptus koraiensis (Eriophyoidea, Phytoptidae) using PacBio long-reads sequencing, Illumina short-reads sequencing, and high-throughput chromatin conformation capture (Hi-C) sequencing. Our assembly resulted in a genome size of 47 Mb across two chromosomes, with scaffold N50 lengths of 24.53 Mb (Table 1). This genome is the first chromosome-level genome among eriophyoid mites, providing significant new data resources for understanding the Eriophyoidea.
Methods
Sample collection
At least 100,000 wild S. koraiensis individuals, including eggs, juveniles and adults, were collected from Pinus koraiensis Siebold & Zucc. (Pinaceae), in Lishui, Nanjing city, Jiangsu province, China (31.3921°N, 118.5417°E). Samples were identified by morphological characteristics with molecular evidence (mitochondrial COI). Vouchers were deposited in the Arthropod/Mite Collection of the Department of Entomology, Nanjing Agricultural University, Jiangsu Province, China.
Genome sequencing
Genomic DNA was extracted from more than 100,000 individuals using MagAttract HMW DNA Kit. The Pacbio 30 kb SMRTbell library was prepared with more than 5 μg gDNA using the SMRTbellTM Prep Kit 2.0 (Pacific Biosciences). The mode of Continuous Long Read (CLR) was run on the Sequel II platform. Illumina whole-genome sequencing was prepared using a 350 bp-insert fragment library (150 bp paired-end) by Truseq DNA PCR-free Kit, which was further sequenced on an Illumina NovaSeq 6000 platform. High-throughput chromosome conformation capture (Hi-C) included cross-linking, HindIII restriction enzyme digestion, end repair, DNA cyclization, purification and capture. The Hi-C library with 300–700 bp insert size library was sequenced on the NovaSeq 6000 platform. Finally, we generated 24.25 Gb (~496X) PacBio long reads, 9.5 Gb (~194X) Illumina short reads, and 9 Gb Hi-C (~184X) reads for our genome assembly.
Genome survey
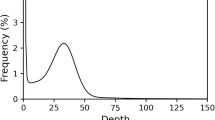
Duplicate and low-quality Illumina raw reads (base quality < Q20, length < 15 bp, polymer A/G/C/ > 10 bp) were trimmed and removed using BBtools package v38.828. The 21-mer depth distribution was counted using script ‘khist.sh’ of BBtools. Genome Scope v2.09 was used to estimate the genome size and heterozygosity of S. koraiensis with the maximum kmer coverage at 1,000×. Based on the distribution of kmer coverage and frequency, the estimated genome size of S. koraiensis was 45.72 Mb, with a heterozygosity rate of around 1.13% and a repeat content proportion of approximately 3.3% (Fig. 1).

GenomeScope genome size estimates for Setoptus koraiensis.
Genome assembly
The CLR reads were set as input to Flye v2.610 to assemble continuous long reads. One round of built-in long reads polishing was performed by Flye v2.6. Then, two rounds of short reads were used to polish and fill in gaps of the primary assembly with NextPolish v1.4.111. Haplotigs and duplication caused by haplotype divergence were eliminated by Purge_dups v1.2.512 using the alignment program Minimap2 v2.2813. Hi-C reads were aligned to the purged genome using BWA v0.7.1814 and Juicer v1.615 to anchor, order and orient contigs into chromosomal assembly following 3D-DNA16 pipeline. Then, we manually reviewed and corrected assembled errors using Juicebox v2.1717. Contaminations were checked and deleted against the UniVec and NCBI nucleotide databases using BLAST + v2.11.018 and MMseqs2 v1619. The completeness of genome assembly was evaluated by BUSCO version 5.2.220 using the eukaryota_odb10 dataset (creation date 2020-09-10). The reads from the whole genome sequencing were aligned back to the genome assembly to access the mapping rate. After de novo assembly, polishing and contaminant removal, the S. koraiensis genome has a genome size of 49.9 Mb with 565 scaffolds, an N50 length of 24.53 Mb, with 94.2% of assembled genomes anchored to two chromosomes (Fig. 2) resulting in a final genome size of 47 Mb (Table 1).

Genome-wide chromosomal heatmap of Setoptus koraiensis, the blue boxes show super scaffolds.
Genome annotation
The repetitive elements were identified using RepeatModeler v2.0.521, which discovered the complete long terminal repeats (LTR) with the ‘-LTRstruct’ pipeline. RepeatMasker v4.1.622 was searched against the custom repeat library of Dfam 3.823 and Repbase v2018102624 with options ‘-no_is -norna -xsmall -q’ to soft mask repeats of the genome assembly.
For gene structure annotation, we performed a pipeline integrating ab initio and homolog-based methods. Braker v2.1.525 was used to obtain ab initio gene predictions employing GeneMark-ES/ET/EP v4.3326 and Augustus v3.4.027 based on reference proteins from the OrthoDB v11 database28. GeMoMa v1.929 was used for homology prediction with the parameters “GeMoMa.c = 0.4 GeMoMa.p = 10”, and the protein sequences of six species (Aculops lycopersici (GCA_015350385.1), Tetranychus urticae (GCA_039701765.1), Tetranychus piercei (GCA_036759885.1), Panonychus citri (GCA_014898815.1), Pyemotes zhonghuajia (GCA_025170145.1), Blomia tropicalis (GCA_029204025.1)) were provided to assist gene prediction. The results obtained from BRAKER and GeMoMa were combined and provided to MAKER v3.01.0330. The functional annotation of predicted protein sequences was searched against UniProt, InterProScan and eggNOG databases. Diamond v2.1.1031 was used to assign the gene function of the best hits in the UniProt database under the ‘very sensitive’ mode. Gene Ontology (GO) and pathway (KEGG) were annotated using InterProScan v5.7232 and eggnog-mapper v2.1.1233 against Pfam34, SMART35, Superfamily36, CDD37, and EggNOG 5.0.2 database38.
Data Records
The raw reads and genome assembly have been deposited in the NCBI databases under BioProject PRJNA1196018. The PacBio, Illumina, and Hi-C data are available under identification numbers SRR32458739-SRR3245874139. The final chromosome assembly has been deposited at GenBank under the accession number GCA_048013815.140. The mitochondrial COI sequence has been deposited at GenBank under the accession number PV16383341. The genome assembly and annotation files are available in Figshare42.
Technical Validation
We mapped the Illumina sequencing data to the final assembly with BWA v0.7.18, and the mapping rate was 92.9%. We assessed the completeness of the genome assembly using BUSCO v5.4.2 with the ‘eukaryota_odb10’ database, and a total of 89% (83.9% single-copied genes, 5.1% duplicated genes, 5.5% fragmented, and 5.5% missing genes) completed BUSCOs were identified, which is higher than that of A. lycopersici (86.3%). We masked 13.82% (6.42 Mb) repetitive regions of the S. koraiensis genome. Among them, 0.2% of repeat sequences were short interspersed elements (SINEs), 1.29% were long interspersed elements (LINEs), 0.92% were long terminal repeats (LTRs), 1.61% were DNA transposons, and 5.14% were unclassified (Fig. 3). We identified 5,954 protein-coding genes, with 4,770 genes that could be functionally annotated. The BUSCO completeness for protein sequence is 77.3% (71.4% single-copied genes, 5.9% duplicated genes, 3.9% fragmented, and 18.8% missing genes) with the ‘eukaryota_odb10’ database. All evidence strongly supported the completeness and accuracy of S. koraiensis genome assembly.

Circular karyotype representation of the chromosomes of Setoptus koraiensis. Tracks from inside to outside are GC content (GC), density of protein-coding genes (GENE), DNA transposons (DNA), LTR/LINE/SINE retrotransposons (LTR, LINE, SINE), and simple repeats (Simple).
Code availability
No custom scripts or code were used in this study.
References
Zhang, Z.-Q. Eriophyoidea and allies: where do they belong? Syst. Appl. Acarol. 22, 1091–1095 (2017).
Zhang, Z.-Q. Phylum Arthropoda von Siebold, 1848. in Animal biodibersity: An Outline of Higher-Level Classification and Survey of Taxonomic Richness (ed. Zhang, Z.-Q.) 99–103 (Magnolia Press, 2011)
Li, N., Sun, J.-T., Yin, Y., Hong, X.-Y. & Xue, X.-F. Global patterns and drivers of herbivorous eriophyoid mite species diversity. J. Biogeogr. 50, 330–340 (2022).
Skoracka, A., Smith, L., Oldfield, G., Cristofaro, M. & Amrine, J. W. Host-plant specificity and specialization in eriophyoid mites and their importance for the use of eriophyoid mites as biocontrol agents of weeds. Exp. Appl. Acarol. 51, 93–113 (2010).
Yin, Y. et al. DNA barcoding uncovers cryptic diversity in minute herbivorous mites (Acari, Eriophyoidea). Mol. Ecol. Resour. 22, 1986–1998 (2022).
de Lillo, E., Pozzebon, A., Valenzano, D. & Duso, C. An intimate relationship between eriophyoid mites and their host plants–a review. Front. Plant. Sci. 9, 1786 (2018).
Greenhalgh, R. et al. Genome streamlining in a minute herbivore that manipulates its host plant. Elife 9 (2020).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2024) (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell. Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell. Syst. 3, 99–101 (2016).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Steinegger, M. & Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2024) (2013–2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic. Acids. Res. 44, D81–89 (2016).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11 (2015).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR. Genom. Bioinform. 3, lqaa108 (2021).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR. Genom. Bioinform. 2, lqaa026 (2020).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic. Acids. Res. 32, W309–312 (2004).
Kuznetsov, D. et al. OrthoDB v11: annotation of orthologs in the widest sampling of organismal diversity. Nucleic. Acids. Res. 51, D445–D451 (2023).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic. Acids. Res. 44, e89 (2016).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic. Acids. Res. 45, D190–D199 (2017).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic. Acids. Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic. Acids. Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic. Acids. Res. 37, D380–386 (2009).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic. Acids. Res. 45, D200–D203 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic. Acids. Res. 47, D309–D314 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP565774 (2024).
Shao, Z.-K. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_048013815.1 (2025).
Shao, Z.-K. GenBank https://identifiers.org/ncbi/insdc:PV163833 (2025).
Shao, Z.-K., Chen, L., Sun, J.-T. & Xue, X.-F. A chromosome-level genome assembly of eriophyoid mite Setoptus koraiensis. figshare https://doi.org/10.6084/m9.figshare.28087958 (2025).
Acknowledgements
This research was funded by the National Natural Science Foundation of China (32170466). This work was also supported by the high-performance computing platform of Bioinformatics Center, Nanjing Agricultural University.
Author information
Authors and Affiliations
Contributions
Z.-K.S. and X.-F.X. conceived and designed the study. Z.-K.S. analyzed the data. X.-F.X., L.C. and J.-T.S. had substantial contributions to the interpretation of the data, writing, and review of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shao, ZK., Chen, L., Sun, JT. et al. A chromosome-level genome assembly of eriophyoid mite Setoptus koraiensis. Sci Data 12, 446 (2025). https://doi.org/10.1038/s41597-025-04814-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04814-2
