
Abstract
The oriental armyworm, Mythimna separata, poses a persistent challenge to agricultural pest management due to its strong migratory abilities and polyphagous feeding behavior. In this study, we present a chromosome-level genome assembly using Illumina, PacBio HiFi, and Hi-C sequencing technologies. The final assembly spans 714.5 Mb with a scaffold N50 of 22.7 Mb and a GC content of 38.8%. A total of 32 chromosomes were successfully anchored, including the Z and W sex chromosomes. BUSCO analysis indicated a genome completeness of 98.6%, and 19,879 protein-coding genes were predicted. The W chromosome, measuring 30.55 Mb with a repeat content of 68.34%, harbors 824 protein-coding genes. Furthermore, a PCR-based method confirmed W-linked sequences for female-specific sex detection via the ZW system. This enhanced genome assembly provides a valuable resource for evolutionary research on M. separata and facilitates the development of sex-regulated pest control strategies.
Similar content being viewed by others

Background & Summary
Advances in sequencing technologies have enabled researchers to gain deeper insights into genome structure, function, and evolution. Despite the availability of some genomic resources for the oriental armyworm (Mythimna separata)1,2, this pest remains a persistent and evolving challenge in agricultural pest management. Renowned for its destructive potential, M. separata causes severe damage to a wide range of crops, including rice, maize, and wheat, across Asia and Australia3,4. It is recognized as one of the most significant polyphagous and invasive insect species.
Global shifts in climate, crop planting patterns, varietal distribution, and cultivation systems have driven M. separata to develop new patterns of adaptability5. These changes have enhanced its ability to thrive in diverse environmental habitats, further intensifying its threat to agricultural systems. The migration behavior of M. separata plays a key role in its outbreaks, enabling its polyphagous impact across vast geographical regions. Its range extends from northern areas, including Korea and Japan, to southern regions such as the Indochina Peninsula, the Philippines, and Malaysia6,7. While limited cold tolerance prevents M. separata from overwintering in most regions, seasonal migrations allow it to reach suitable areas for survival and reproduction8,9. These biological traits highlight the importance of M. separata as a critical subject for in-depth research.
In this study, we present an enhanced chromosome-level 714.5 Mb genome assembly for M. separata, including the complete assembly of both sex chromosomes, featuring the longest and most complete W chromosome reported for this species to date. This genome version offers high sequence accuracy and improved contiguity. Additionally, we developed markers for sex detection based on W chromosome-specific sequences, enabling the identification of M. separata’s sex even at the larval stage. By addressing gaps in existing genomic data, this improved assembly provides a robust foundation for understanding the genetic architecture underlying M. separata’s environmental adaptability and offers valuable resources for advancing pest management strategies.
Methods
Sample information
The Mythimna separata samples used in this study were derived from an inbred strain reared on a noctuid artificial diet under controlled laboratory conditions (25 ± 2 °C, 14L:10D photoperiod, and 75 ± 5% relative humidity) at the Agricultural Genomics Institute of Chinese academy of Agricultural Sciences at Shenzhen. A single-pair mating approach was employed to produce subsequent generations, followed by consistent sibling mating to ensure genetic homozygosity. After four successive generations, one female pupa was selected for genomic sequencing using PacBio HiFi and Illumina technologies, while another female pupa from the same generation was chosen for Hi-C sequencing. Additionally, individuals from the same generation, representing various developmental stages, were collected for transcriptome sequencing (Table 1). These included larvae at the 2nd, 3rd, 4th, and 5th instars, as well as one male and one female pupa, and one male and one female adult.
Genome sequencing and assembly
Genomic DNA (gDNA) was extracted using the DNeasy Blood & Tissue Kit (Cat. no. 69506, Qiagen). The quality and quantity of the gDNA were assessed using a NanoDrop One UV-Vis spectrophotometer (Thermo Fisher Scientific) and a Qubit 3.0 Fluorometer (Invitrogen), following the manufacturers’ protocols. Approximately 0.5 μg of gDNA was used to generate a PCR-free Illumina genomic library with a 350 bp insert size, prepared using the TruSeq Nano DNA HT Sample Preparation Kit (Illumina, USA). This library was sequenced in 2 × 150 bp format on the Illumina NovaSeq 6000 platform, generating 46.59 Gb of raw Illumina data, achieving a 68.41× sequence coverage of the estimated M. separata genome (Table 1). After quality control with fastp (v0.20.1)10 using default parameters, the clean reads were utilized to construct a 17-mer frequency distribution map using Jellyfish (v2.3.1)11. The genome size of M. separata was subsequently estimated to be 681 Mb using GenomeScope (v1.0)12.
Additionally, 5 μg of gDNA from the same individual was used to create ~15 kb SMRTbell insert libraries, which were sequenced on the PacBio Sequel II system (Pacific Biosciences, USA). A total of 38.71 Gb of clean PacBio HiFi reads was generated, representing 56.89× coverage of the estimated genome (Table 1). The PacBio HiFi reads were then assembled using Hifiasm (v0.3.0)13 with default parameters. Haplotigs were removed using Purge_Dups (v1.2.3)14. The initial genome was assembled into 121 contigs with a total size of 714.46 Mb. The contig N50 was measured 20.45 Mb and the maximum contig length was 25.71 Mb.
Hi-C sequencing and chromosome scaffolding
Hi-C library construction was carried out using the same female pupa used for DNA extraction. The DNA was cross-linked in situ, extracted, and digested with the restriction enzyme DpnII. The resulting fragments were ligated to form chimeric junctions, followed by purification and amplification of the Hi-C libraries using 12–14 cycles of PCR. The libraries were sequenced on the Illumina NovaSeq 6000 platform using a 150 bp paired-end configuration, producing 64.01 Gb of Hi-C read data.
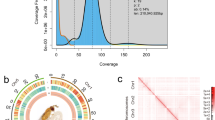
Low-quality raw reads were removed using fastp v0.20.110 with default parameters. The purified reads were then mapped to the assembled contigs using Juicer (v1.5)15. The 3D-DNA pipeline16 was employed for contig clustering, ordering, and phasing, resulting in preliminary chromosomal scaffolds. The chromosome interaction matrix was further manually adjusted using JuiceBox v1.11.0817 at a resolution of 500 kb (Fig. 1). Through Hi-C scaffolding, the final genome assembly measured 714.5 Mb, with a scaffold N50 of 22.7 Mb and a GC content of 38.8%. A total of 32 pseudochromosomes were obtained, representing 97.2% of the total genome size.

Chromosomal architecture of M. separata. (a) Hi-C contact heatmap displaying genomewide chromatin interactions across 32 pseudochromosomes. The intensity along the diagonal patches reflects the frequency of physical interactions between chromatin regions. (b) Karyotypic analysis of the diploid chromosome complement, illustrating a total of 62 chromosomes.
To identify the sex chromosomes from the assembled pseudochromosomes, a synteny analysis was performed by comparing the Mythimna separata genome with those of Spodoptera litura (GCA_002706865.1) and Noctua pronuba (GCA_905220335.1). BLAST v2.226+ was used with a stringent E-value cutoff of <1E−10 to align the amino acid sequences of protein-coding genes from these species. Syntenic blocks were then constructed using MCScanX18 with default parameters, and the relationships were visualized using the TBtools (v2.008)19 package. The analysis revealed that Chr1 corresponds to the Z chromosome, with a length of 34.92 Mb, while Chr2 corresponds to the W chromosome, with a length of 30.55 Mb (Fig. 2). This represents the longest and most accurate W chromosome sequence reported to date (Table 2).

Syntenic relationships between chromosomes of M. separata and those of S. litura (a) and N. pronuba (b). The sex chromosomes, W and Z, are highlighted in red text for each species. Notably, the W chromosome of M. separata lacks a corresponding counterpart in S. litura but aligns with a chromosome in N. pronuba.
RNA sequencing and genome annotation
Total RNA was extracted using the TransZol Up Plus RNA Kit (Cat. no. ER501-01-V2, TransGen), following the manufacturer’s protocol. The integrity and purity of the RNA were evaluated using the same methods employed for gDNA quality assessment, including a NanoDrop One UV-Vis spectrophotometer and a Qubit 3.0 Fluorometer. Indexed cDNA libraries were constructed from the extracted RNA using the NEBNext Ultra RNA Library Prep Kit for Illumina. Libraries with insert sizes of 250–300 bp were sequenced on the Illumina NovaSeq 6000 platform with a paired-end 150 bp strategy, yielding a total of 64.01 Gb of sequencing data.
Several comprehensive approaches were utilized to predict and annotate the genomic features of the M. separata genome. The repeat element identification followed a two-step process. First, a de novo repeat library was constructed using RepeatModeler (v1.0.11)20 based on the assembled genome, with default parameters. Repeat elements were then identified through homology searches against this library using RepeatMasker (v4.07) (https://www.repeatmasker.org/). Transposable element (TE) sequences, including SINEs, LINEs, LTR elements, DNA transposons, and other types, were categorized across the chromosomes. The results of repeat sequence annotation indicate that the W chromosome contains the highest proportion of repetitive sequences, with a particularly significant enrichment of LTR (long terminal repeat) elements compared to the other chromosomes (Table 3).
Gene model prediction for the M. separata genome employed a multi-approach strategy, integrating ab initio, transcriptome-based, and homology-based methods. Initially, the genome was processed with RepeatMasker for masking to minimize interference from repetitive elements. Ab initio gene prediction was carried out using AUGUSTUS (v3.2.2)21 with default parameters, where the model was trained using RNA-Seq-derived transcripts to enhance prediction accuracy. Quality-controlled RNA-Seq reads were aligned to the assembled M. separata genome using TopHat2 (v2.0.12)22 and processed with Cufflinks (v2.2.1)23 to generate transcript predictions. For homology-based prediction, the genome sequences of M. separata were aligned with the protein sequences of closely related species (Agrotis ipsilon, Spodoptera litura, Helicoverpa armigera, Bombyx mori, and Ostrinia furnacalis) using BLAST. Gene structures were subsequently refined and predicted using GeneWise v2.4.1 (https://www.ebi.ac.uk/~birney/wise2/). Finally, Evidence Modeler (v1.1.1)24 was utilized to integrate the predictions from the three approaches, resulting in a unified dataset of 19,879 predicted protein-coding genes. Notably, 824 protein-coding genes were predicted on the W chromosome.
For non-coding RNA (ncRNA) annotation, both database searches and model-based predictions were employed. Transfer RNAs (tRNAs) were identified with tRNAscan-SE v2.0.925 using eukaryote-specific parameters. Ribosomal RNAs (rRNAs) and their subunits were predicted using Barrnap v0.9 (https://github.com/tseemann/barrnap) with the parameter–kingdom euk. MicroRNAs (miRNAs), small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs) were detected by querying the Rfam database (v14.10)26 with Infernal cmscan v1.1.427. This comprehensive analysis identified 4,367 tRNAs, 551 rRNAs, 71 miRNAs, 139 snRNAs, and 23 snoRNAs in the M. separata genome (Table 4).
Development of W-chromosome specific markers
In Lepidoptera, females possess a ZW sex-determination system, whereas males have a ZZ system28,29,30. This results in a female-specific presence of W-linked sequences, which can be used for sex detection, particularly in cases where both sexes share similar morphological characteristics, such as during the larval stage. To identify W-chromosome-specific sequences, we compared them with other chromosomes and designed two pairs of PCR primers accordingly (Table 5). PCR amplification and agarose gel electrophoresis revealed that specific bands corresponding to the W chromosome were present exclusively in females, as expected in the ZW sex-determination system of Lepidoptera species (Fig. 3). This sex-specific banding pattern, combined with the positive identification of a mitochondrial COI marker31, enabled clear and reliable detection of M. separata’s sex, even at the larval stage.

Sex detection markers based on W chromosome in M. separata. (a) Schematic representation of the sex detection method using PCR amplification and gel electrophoresis, where W-chromosome-specific bands are observed exclusively in females. (b) Electrophoresis results for two developed sex markers, with a mitochondrial COI marker included as a positive control. Lanes 1–10 correspond to male samples, and lanes 11–20 correspond to female samples. Notably, no bands were detected in lane 16, likely due to poor DNA quality.
Data Records
The whole genome project is accessible in NCBI’s database under BioProject accession number PRJNA1194333. The PacBio, Illumina, Hi-C sequencing and RNA-Seq raw data are available on the NCBI Sequence Read Archive under accession number SRP55021832. The assembled genome is publicly available in GenBank under accession number GCA_048418785.133. To enhance accessibility, the assembled genome and gene annotation files have been deposited in the Figshare database34.
Technical Validation
To ensure the integrity and accuracy of the assembled chromosomes, several validation approaches were undertaken. First, karyotyping was performed to determine the chromosome number and configurations of M. separata. For karyotype analysis, testis was dissected from a 5th instar larva and incubated in a colchicine solution at 25 °C for 3 hours. The tissues underwent hypotonic treatment in 1% sodium citrate solution at 25 °C for 50 minutes, followed by fixation in a 3:1 methanol–acetic acid solution at 4 °C for 2 hours. Next, the tissues were softened in 60% acetic acid for 10 minutes on pre-chilled glass slides, fully shredded using a dissecting needle, and refixed in the same fixative to create an even cell suspension. After air-drying, the slides were stained with 5% Giemsa solution (pH 6.8) for 30 minutes, rinsed with running water, and air-dried at room temperature. Chromosome morphology was observed using an Olympus BX51 microscope. The analysis revealed that the diploid complement of M. separata consists of 62 chromosomes, providing critical insights into the chromosomal architecture of this species (Fig. 1).
To evaluate assembly integrity, PacBio HiFi sequencing reads were mapped to the assembled genome using the minimap2 tool35. The alignment analysis demonstrated an average sequencing depth of 51.33× and a coverage rate of 99.99%, indicating near-complete genome representation. The completeness of the M. separata genome assembly was further evaluated using BUSCO (v5.3.2)36 with the Arthropoda gene set (odb10). The results showed that 98.6% of BUSCO genes were complete, comprising 97.1% single-copy and 1.5% duplicated genes, with 0.2% fragmented and 1.2% missing BUSCOs. Similarly, a quality assessment of the predicted gene set in protein mode revealed a BUSCO completeness of 97.8%, including 96.3% single-copy, 1.5% duplicated, 0.7% fragmented, and 1.5% missing BUSCOs. These metrics collectively affirm the assembly’s high quality and completeness.
To further validate the chromosomal organization, the raw Illumina reads were mapped to the 32 chromosomes, revealing that the sequencing depth of the W and Z chromosomes is nearly half that of the autosomes (Table 3). This is consistent with the female pattern, where the sex chromosomes are haploid, while the autosomes are diploid. Additionally, the synteny analysis demonstrated strong syntenic relationships between the M. separata Z chromosome and the Z chromosomes of the other two species (Fig. 2). In contrast, the W chromosome of M. separata exhibited high synteny with the W chromosome of N. pronuba37, which is notably absent in S. litura38 (Fig. 2). These results confirm the identity and presence of the W sex chromosome in this current genome assembly.
Code availability
No custom scripts were utilized in this study. All bioinformatics analyses were conducted using standard protocols as outlined in the documentation of the respective tools and software. The software versions and parameters used are detailed throughout the manuscript.
References
Xu, C. et al. Chromosome level genome assembly of oriental armyworm Mythimna separata. Scientific Data 10, 597, https://doi.org/10.1038/s41597-023-02506-3 (2023).
Zhao, H. et al. Chromosome-level genomes of two armyworms, Mythimna separata and Mythimna loreyi, provide insights into the biosynthesis and reception of sex pheromones. Molecular Ecology Resources 23, 1423–1441, https://doi.org/10.1111/1755-0998.13809 (2023).
Li, B.-L., Li, M.-M., Li, T.-T., Wu, J.-X. & Xu, X.-L. Demography of Mythimna separata (Lepidoptera: Noctuidae) at outdoor fluctuating temperatures. Bulletin of Entomological Research 111, 385–393, https://doi.org/10.1017/S0007485321000110 (2021).
Tong, D. et al. The oriental armyworm genome yields insights into the long-distance migration of noctuid moths. Cell Rep 41, 111843, https://doi.org/10.1016/j.celrep.2022.111843 (2022).
Liu, Y. et al. Chitin deacetylase: A potential target for Mythimna separata (Walker) control. Arch Insect Biochem Physiol 104, e21666, https://doi.org/10.1002/arch.21666 (2020).
Jiang, X. et al. Biocontrol of the oriental armyworm, Mythimna separata, by the tachinid fly Exorista civilis is synergized by Cry1Ab protoxin. Scientific Reports 6, 26873, https://doi.org/10.1038/srep26873 (2016).
Sharma, H. C. & Davies, J. C. The oriental armyworm, Mythimna separata (Wlk.) distribution, biology and control: a literature review. Miscellaneous Reports-Centre for Overseas Pest Research 59, 1–24 (1983).
Ali, A., Rashid, M. A., Huang, Q. Y., Wong, C. & Lei, C. L. Response of antioxidant enzymes in Mythimna separata (Lepidoptera: Noctuidae) exposed to thermal stress. Bull Entomol Res 107, 382–390, https://doi.org/10.1017/s0007485316001000 (2017).
Izadi, H., Cuthbert, R. N., Haubrock, P. J. & Renault, D. Advances in understanding Lepidoptera cold tolerance. Journal of Thermal Biology 125, 103992, https://doi.org/10.1016/j.jtherbio.2024.103992 (2024).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3, 99–101 (2016).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
Chen, C. et al. TBtools-II: A “One for All, All for One” Bioinformatics Platform for Biological Big-data Mining. Mol Plant https://doi.org/10.1016/j.molp.2023.09.010 (2023).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36 (2013).
Ghosh, S. & Chan, C. K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol Biol 1374, 339–361 (2016).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol Biol 1962, 1–14, https://doi.org/10.1007/978-1-4939-9173-0_1 (2019).
Ontiveros-Palacios, N. et al. Rfam 15: RNA families database in 2025. Nucleic Acids Research 53, D258–D267, https://doi.org/10.1093/nar/gkae1023 (2024).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337, https://doi.org/10.1093/bioinformatics/btp157 (2009).
Traut, W., Sahara, K. & Marec, F. Sex chromosomes and sex determination in Lepidoptera. Sex Dev 1, 332–346, https://doi.org/10.1159/000111765 (2007).
Fraïsse, C., Picard, M. A. L. & Vicoso, B. The deep conservation of the Lepidoptera Z chromosome suggests a non-canonical origin of the W. Nature Communications 8, 1486, https://doi.org/10.1038/s41467-017-01663-5 (2017).
Nagaraju, J., Gopinath, G., Sharma, V. & Shukla, J. N. Lepidopteran sex determination: a cascade of surprises. Sex Dev 8, 104–112, https://doi.org/10.1159/000357483 (2014).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3, 294–299 (1994).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP550218S (2024).
Zhang, L., Yesaya, A. & Liang, X. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_048418785.1 (2025).
Zhang, L., Yesaya, A., Wu, C. & Xiao, Y. Chromosomal-level genome dataset of Mythimna separata.figshare https://doi.org/10.6084/m9.figshare.27958434.v4 (2024).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 1962, 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Boyes, D. & Holland, P. W. H. The genome sequence of the large yellow underwing, Noctua pronuba (Linnaeus, 1758). Wellcome Open Res 7, 119, https://doi.org/10.12688/wellcomeopenres.17747.1 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_002706865.3 (2017).
Acknowledgements
The research was financially supported by the STI 2030–Major Projects (Grant No. 2022ZD04021) and the Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences (Grant No. CAASZDRW202412).
Author information
Authors and Affiliations
Contributions
Y.X. and L.Z. conceptualized and led the project; L.Z. and X.L. prepared the samples for sequencing. L.Z. and X.L. performed lab experiments for sex detection including designing the markers; L.Z., A.Y. and Z.L. contributed to genome assembly, annotation, data visualization, and other bioinformatics analyses. L.Z. and A.Y. drafted and wrote the manuscript, while all authors reviewed, revised, and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, L., Yesaya, A., Li, Z. et al. A high-quality chromosome-level genome assembly for the agricultural pest Mythimna separata. Sci Data 12, 540 (2025). https://doi.org/10.1038/s41597-025-04855-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04855-7
This article is cited by
-
A chromosome-level genome assembly of the Hermonassa cecilia (Lepidoptera: Noctuidae)
Scientific Data (2025)
