
Abstract
The genus Helianthemum, commonly known as rockroses, encompasses 140 species primarily distributed in the Palearctic region, with notable diversification driven by climatic and geological changes. These plants are valuable for studying speciation processes and ecological divergence. The chemical properties of the leaves have also been investigated for containing valuable bioactive compounds with several therapeutic properties. However, the availability of genomic resources for species in this genus are almost entirely lacking. Here, we assembled and annotated the first reference transcriptome of Helianthemum marifolium, a species with wide morphological variability and infraspecific diversity. Illumina paired-end RNA sequences were generated using leaves from 16 individuals, representing the four recognized subspecies, all cultivated in a greenhouse. RNA reads were assembled with Trinity and Oases, and EvidentialGene produced a transcriptome with 122,002 transcripts. The transcriptome showed 59524 hits on the UniProtBK database through BLASTx. This transcriptome will be an invaluable resource for transcriptome-level population studies, conservation genetics of the many endangered species within the genus, and for deepen into the metabolic pathways of leaf-derived compounds.
Similar content being viewed by others


Background & Summary
The family Cistaceae comprises eight genera and 180 species commonly known as rockroses, with numerous taxa of considerable biochemical, biological, and ecological importance1,2. Helianthemum Mill. is the largest genus in this family, forming a monophyletic, complex, and species-rich Palearctic plant lineage with approximately 140 species and subspecies, ranging from Macaronesia to Central Asia3,4. The rapid evolutionary diversification of Helianthemum is centred in the Mediterranean region and has been driven by major paleoclimatic and geological events, especially since the Upper Miocene5. Ecological divergence has played a key role in promoting reproductive isolation and driving phenotypic differences between lineages within this genus4,6. Nowadays Helianthemum species can grow under severe aridity conditions in deserts, alpine pastures, and Mediterranean maquis, where they are exposed to drastic environmental conditions. It is not surprising that several lines of evidence point to Helianthemum as an attractive model system for studying incipient speciation and the evolution of species complexes. These complexes are characterized by a notable degree of intraspecific taxonomic diversity, primarily manifested in vegetative differentiation. Among the most significant taxonomic characters are those related to leaf morphology, particularly the presence, shape, type, and abundance of trichomes7. Additionally, Helianthemum leaves are a natural source of compounds with biological, aromatic, and pharmacological properties, showing a taxon-dependent variation in the chemical profile8. The leaves are rich in phenolics and flavonoids, which regulate the expression of various cytoprotective genes against inflammation and oxidative stress, and contain secondary metabolites with antiparasitic, antibacterial, and antifungal activity9,10,11,12,13.
Differential gene expression can evolve rapidly and become the basis for ecological divergence14,15. Therefore, studying expression divergence may help elucidate phenotypic differences observed even among recently diverged lineages16,17. However, the metabolic pathways of leaf compounds in Helianthemum have never been studied using a transcriptomic approach. Despite its ecological and biochemical significance, as well as its suitability as a model for studying niche adaptation and speciation, extraordinarily few genomic resources are available for Helianthemum18. No transcript sequence information for leaf tissues in the genus Helianthemum is readily available, and the only existing de novo transcriptome assembly within this genus is limited to root tissues of H. almeriense artificially inoculated with mycorrhizae19.
In this study, we report the first de novo assembled transcriptome of Helianthemum leaves, specifically for H. marifolium (L.) Mill., a species that is geographically restricted to the south and east of the Iberian Peninsula and southern France. Phylogenomic data support the existence of four recognised subspecies within this species, which exhibit intricate morphological variations with a genetic basis, potentially associated with environmental adaptation4,20. The transcriptome assembled in this study is a helpful resource for functional genomics and the elucidation of molecular processes, as well as for studying differentially expressed genes involved in both macro- and micro-evolution and local adaptation. Moreover, it offers valuable molecular resources. For instance, the transcriptome developed here can be mined for the identification of EST-SSR markers, which can be applied to assess genetic variation patterns in the numerous threatened and critically endangered species within the genus Helianthemum. This resource may prove invaluable for the development of effective conservation action plans. Finally, this reference transcriptome will facilitate integrated transcriptome and metabolome analyses, assisting researchers in the analysis of functional genes, including those involved in the taxa-dependent biosynthesis of important secondary metabolites.
Methods
Sample collection
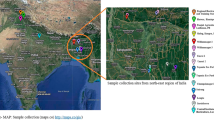
Seeds of the four subspecies of Helianthemum marifolium (andalusicum, marifolium, molle and origanifolium) were collected in Spain from eight wild populations (two populations for each subspecies; Table 1). The use of multiple subspecies is suitable to address the physiological variability observed within a species. Indeed, some of these infraspecific taxa possess notable ethnopharmacological value, containing flavonoids and exhibiting a higher polyphenol content than other Helianthemum species, with a probable subspecies-dependent profile8,21.
The seeds were subjected to mechanical scarification with fine-grained sandpaper and germinated on sterile Petri plates with moist filter paper. Five days later, the seedlings were transferred to soil-filled pots containing a 3:1:1 ratio of soil:sand:perlite, and cultivated in a greenhouse at 22/25°C (night/day), 40–60% relative humidity, and natural daylight. At 446 days after sowing, two adult plants from each population of origin (i.e., four replicates of every subspecies, 16 plants in total) were selected for RNA isolation. The leaves from the 3–5 apical nodes were frozen immediately in liquid nitrogen and stored at −80 °C until processing at Novogene Company (UK, Cambridge).
RNA extraction, RNA-seq library construction and sequencing
The RNA was extracted using the RNeasy Plant Mini Kit (QIAGEN, Crawley, UK), following the manufacturer’s instructions. Messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. Following fragmentation, the first-strand cDNA was synthesised using random hexamer primers followed by the second-strand cDNA synthesis. The library was prepared following the steps of end repair, A-tailing, adapter ligation, size selection, amplification, and purification. The Novogene NGS RNA Library Prep Set was employed for library preparation.
The library was evaluated using Qubit and real-time PCR for quantification and with the Agilent 5400 Fragment Analyzer system (Agilent, USA) for size distribution detection. The quantified libraries were subsequently pooled, and RNA sequencing was conducted using an Illumina platform (Novaseq 6000), resulting in the generation of paired-end 150 bp reads.
RNA-Seq read quality control and cleaning
An overview of the bioinformatic workflow is shown in Fig. 1. The raw sequence data were subjected to quality control checks using FastQC v0.12.1 software. Erroneous K-mers were corrected with Rcorrector v1.0.722 and paired-end reads were trimmed using Fastp v1.0.723 to remove adapters, with a Phred quality score of 20 selected. An additional cleaning step was undertaken to remove rRNA using SortMeRNA v4.3.624 with the mr_v4.3 database serving as the reference. Finally, potential contaminants were removed with kraken2 v2.1.325 using the PlusPF and nt databases (see also Technical Validation section).

Flowchart illustrating the bioinformatic steps and tools used for analysis, from quality trimming and filtering to transcriptome assembly and annotation.
Normalization, de novo transcriptome assembly and annotation
The cleaned read files of the 16 sampled individuals were concatenated into single files for the forward and reverse reads, respectively, and normalized in silico with the Perl script InSilico_read_normalization.pl from Trinity v2.8.626. The normalized files were subjected to five different assemblies, one with Trinity and four with Oases v0.2.827 with different k-mers (25, 31, 41, 51). All the assemblies were merged into a single file and subjected to the tr2aacds.pl pipeline of EvidentialGene v2023.07.1528 which is designed to obtain the optimal biologically useful “best” set of mRNAs (non-redundant, containing the best-assembled transcripts from each assembler) from several assemblies performed by different methods. For this study, only the “okay set” was selected for further analysis.
The annotation was carried out using TransDecoder v5.7.1 (https://github.com/TransDecoder/TransDecoder)29 to predict candidate coding regions. BLASTx v2.5.0 was used for the similarity search based on the homology of transcripts and predicted proteins with the latest version of the UniProtKB/Swiss-Prot database. The search was conducted with a maximum E-value threshold of 1e-5 and 59524 transcripts returned a positive BLAST hit against the database. The Gene Ontology (GO) annotations were performed using Trinotate v4.0.230, resulting in 44188 transcripts assigned to different GO terms related to the three primary categories: cellular component, molecular function, and biological process. The most abundant GO terms annotated are shown in Fig. 2.

Bar chart of the most frequent (top 10) Gene Ontology (GO) annotated terms associated to the obtained transcripts, corresponding to the different GO categories (a) transcripts assigned with GO terms in Cellular Component, (b) transcripts in Molecular Function, and (c) transcripts in Biological Process. Each category is sub-categorized in different GO terms, represented on y-axis and numbers of transcripts are shown in x-axis.
Mapping, count table, and expression analyses
The software Salmon v1.10.031 was used to map reads against the reference transcriptome and generate equivalence classes for reads from each sample. Corset v1.0932 was employed to obtain gene-level counts rather than transcript-level counts, resulting in a count matrix. This was then imported into R for expression analyses, while a file containing the clusters was used for gene annotation.
The count data were input into edgeR v4.4.033 for filtering and normalisation using the TMM method. Exploratory analyses were then performed using FactoMineR v2.1134, including a principal component analysis (PCA) and a cluster analysis on the normalised counts. Preliminary expression results obtained with the PCA of gene expression and the hierarchical clustering of samples revealed a well-defined separation of the four subspecies, and a similar gene expression pattern within subspecies (Fig. 3). The transcriptome presented here adds a valuable resource for comparative genomics studies in the genus Helianthemum expanding molecular data for evolutionary studies in the family Cistaceae and can be useful to uncover single nucleotide polymorphisms (SNPs) in the coding regions of the genome. The accurate transcript annotation will enable us to figure out the gene function of particular traits of interest and expression profiling, providing indispensable transcriptomic resources for future studies on the cell signalling pathways.

Similarity analysis of RNA-seq samples. (a) Principal component analysis of gene expression, with samples from different subspecies highlighted in different colours. (b) Hierarchical clustering of samples, with each subspecies’ samples highlighted in different colours. The numbers in the labels of each sample represent the population code (see Table 1) followed by the individual identifier.
Data Records
The raw reads of the 16 samples used for the transcriptome assembly are deposited in the NCBI Sequence Read Archive (SRA) database with accession number SRP52272735. The assembled and curated transcriptome, the predicted peptide sequences from TransDecoder and the gene ontology annotations from Trinotate can all be found in the Zenodo public repository36. The raw gene-level count matrix and the assembled transcriptome are also available at the Gene Expression Omnibus (GEO) database under accession GSE29193537.
Technical Validation
Assembly quality control
Read quality assessment was conducted using FastQC. Transcriptome assembly validation and completeness were performed using Benchmark Universal Single-Copy Orthologs (BUSCO) v5.7.038 and Quast v5.0.239. Our final assembled transcriptome had a total of 122,002 contigs (of which the largest size was 15,683 bp), a N50 of 1533 bp (Table 2), and 88.4% of the 2805 genes corresponding to BUSCOs database eudicotyledons_odb12 (Completeness: 88.4% [Single copy: 69.4%, Duplicates: 19.0%], Fragmented: 5.8%, Missing: 5.8%).
Removal of contaminant reads prior to transcriptome assembly is an important step to ensure the quality of the delivered transcriptome. In our study, of the 487,286,173 forward sequences processed with kraken225, 14,978,328 were classified and eliminated with the first (PlusPF) database, and 1,005,362 with the second (nt) database, retaining a total of 471,302,483 sequences for normalization. As for the 487,086,862 reverse sequences, 14,997,325 were classified and eliminated with the first database, and 998,892 with the second, leaving a total of 471,090,645 sequences for normalization.
Code availability
All code used in this study is available at https://github.com/Andreamadi.
References
Papaefthimiou, D. et al. Genus Cistus: a model for exploring labdane-type diterpenes’ biosynthesis and a natural source of high value products with biological, aromatic, and pharmacological properties. Front. Chem. 2, 35 (2014).
Papanikolaou, A. S. et al. Chemical and transcriptomic analyses of leaf trichomes from Cistus creticus subsp. creticus reveal the biosynthetic pathways of certain labdane-type diterpenoids and their acetylated forms. J. Exp. Bot. 75, 3431–3451 (2024).
Aparicio, A. et al. Phylogenetic reconstruction of the genus Helianthemum (Cistaceae) using plastid and nuclear DNA-sequences: Systematic and evolutionary inferences. Taxon 66, 868–885 (2017).
Martín-Hernanz, S. et al. Maximize resolution or minimize error? Using genotyping-by-sequencing to investigate the recent diversification of Helianthemum (Cistaceae). Front. Plant Sci. 10, 1416 (2019).
Martín-Hernanz, S. et al. Biogeographic history and environmental niche evolution in the Palearctic genus Helianthemum (Cistaceae). Mol. Phylogenet. Evol. 163, 107238 (2021).
Martín-Hernanz, S. et al. Strong conservatism of floral morphology during the rapid diversification of the genus Helianthemum. Am J Bot. 110, e16155 (2023).
Widén, B. Inheritance of a hair character in Helianthemum oelandicum var. canescens and allele frequencies in natural populations. Plant Syst. Evol. 304, 145–161 (2018).
Rubio-Moraga, Á. et al. Screening for polyphenols, antioxidant and antimicrobial activities of extracts from eleven Helianthemum taxa (Cistaceae) used in folk medicine in south-eastern Spain. J. Ethnopharmacol. 148, 287–296 (2013).
Alsabri, S. G. et al. Phytochemical, anti-oxidant, anti-microbial, anti-inflammatory and anti-ulcer properties of Helianthemum lippii. J. Pharmacogn. Phytochem. 2, 86–96 (2013).
Benabdelaziz, I., Marcourt, L., Benkhaled, M., Wolfender, J. L. & Haba, H. Antioxidant and antibacterial activities and polyphenolic constituents of Helianthemum sessiliflorum Pers. Nat. Prod. Res. 31, 686–690 (2017).
Agostini, M. et al. Phytochemical and biological investigation of Helianthemum nummularium, a high-altitude growing alpine plant overrepresented in ungulates diets. Planta Med. 86, 1185–1190 (2020).
Mouffouk, S., Mouffouk, C., Mouffouk, S. & Haba, H. Medicinal, Pharmacological and Biochemical Progress on the Study of Genus Helianthemum: A Review. Curr. Chem. Biol. 17, 147–159 (2023).
Laib, I. et al. Therapeutic potential of silver nanoparticles from Helianthemum lippii extract for mitigating cadmium-induced hepatotoxicity: liver function parameters, oxidative stress, and histopathology in wistar rats. Front. Bioeng. Biotechnol. 12, 1400542 (2024).
Abzhanov, A. et al. The calmodulin pathway and evolution of elongated beak morphology in Darwin’s finches. Nature 442, 563–567 (2006).
Wray, G. A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8, 206–216 (2007).
Chapman, M. A., Hiscock, S. J. & Filatov, D. A. Genomic divergence during speciation driven by adaptation to altitude. Mol. Biol. Evol. 30, 2553–2567 (2013).
Dunning, L. T. et al. Ecological speciation in sympatric palms: 1. Gene expression, selection and pleiotropy. J. Evol. Biol. 29, 1472–1487 (2016).
Duan, Y., Du, Z. & Wang, J. Complete chloroplast genome characteristics of the endangered relic plant Helianthemum songaricum (Cistaceae) in the arid region of northwestern China. Mitochondrial DNA Part B 4, 1961–1962 (2019).
Marqués-Gálvez, J. E. et al. Desert truffle genomes reveal their reproductive modes and new insights into plant–fungal interaction and ectendomycorrhizal lifestyle. New Phytol. 229, 2917–2932 (2021).
Martín-Hernanz, S., Velayos, M., Albaladejo, R. G. & Aparicio, A. Systematic implications from a robust phylogenetic reconstruction of the genus Helianthemum (Cistaceae) based on genotyping-by-sequencing (GBS) data. Anales Jard. Bot. Madr. 78, e113 (2021).
Pardo de Santayana, M. et al. (eds.) Inventario español de los conocimientos tradicionales relativos a la biodiversidad. Fase II. Ministerio de Agricultura y Pesca, Alimentación y Medio Ambiente (2018).
Song, L. & Florea, L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Gigascience 4, (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Schulz, M. H., Zerbino, D. R., Vingron, M. & Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092 (2012).
Gilbert, D. Gene-omes built from mRNA seq not genome DNA. F1000Research 5, (2013).
TransDecoder. GitHub - TransDecoder/TransDecoder: TransDecoder source. GitHub https://github.com/TransDecoder/TransDecoder.
Bryant, D. M. et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 18, 762–776 (2017).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Davidson, N. & Oshlack, A. Data From “Corset: Enabling Differential Gene Expression Analysis For De Novo Assembled Transcriptomes.” Zenodo (CERN European Organization for Nuclear Research) https://zenodo.org/record/11134 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Lê, S., Josse, J. & Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 25, (2008).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP522727 (2024).
Albaladejo, R. G., Martín-Díaz, A., de Vega, C., Martín-Hernanz, S. & Aparicio, A. De novo transcriptome assembly of the rockrose Helianthemum marifolium. Zenodo https://doi.org/10.5281/zenodo.13710166 (2025).
NCBI Gene Expression Omnibus https://identifiers.org/geo/GSE291935 (2025).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Acknowledgements
We are grateful to Encarnación Rubio and the personnel of the CITIUS Greenhouse Service at the Universidad de Sevilla for their assistance during the cultivation of plants and to the staff of the CICA supercomputer for their guidance in using the High-Performance Computing (HPC) facility. This work was supported by grant PID2020-116355GB-I00 from the Spanish Ministerio de Ciencia e Innovación to R.G.A.
Author information
Authors and Affiliations
Contributions
The experiment was conceived by R.G.A., A.A.M. and S.M.H. R.G.A. and C.d.V. conducted the greenhouse experiment and processed the samples. A.M.D. and R.G.A. conducted the bioinformatic analyses. A.M.D., R.G.A. and C.d.V. prepared the manuscript. All authors contributed for reviewing and revising the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Martín-Díaz, A., de Vega, C., Martín-Hernanz, S. et al. De novo transcriptome assembly of the plant Helianthemum marifolium for the study of adaptive mechanisms. Sci Data 12, 515 (2025). https://doi.org/10.1038/s41597-025-04888-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04888-y
