
Abstract
Diagnosing lung cancer at a curable stage offers the opportunity for a favorable prognosis. The emerging epigenomics analysis on plasma cell-free DNA (cfDNA), including 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) modifications, has acted as a promising approach facilitating the identification of lung cancer. And, integrating 5mC biomarker with chest computed tomography (CT) image features could optimize the diagnosis of lung cancer, exceeding the performance of models built on single feature. However, the clinical applicability of integrated markers might be limited by the potential risk of overfitting due to small sample size. Hence, we prospectively collected peripheral blood sample and the paired chest CT images of 2032 patients with indeterminate pulmonary nodules across 5 centers, and constructed a large-scale, multi-institutional, multiomics database that encompass CT imaging data and plasma cfDNA fragmentomic in 5mC-, 5hmC-enriched regions. To our best knowledge, this dataset is the first radio-epigenomic dataset with the largest sample size, and provides multi-dimensional insights for early diagnosis of lung cancer, facilitating the individuated management for lung cancer.
Similar content being viewed by others


Background & Summary
Lung cancer is still the leading cause of death from cancer worldwide1,2, and approximately 75% of lung cancer patients were diagnosed at advanced stage of III-IV. Compared with the increasing 5-year survival rates of stage I lung cancer, the 5-year survival rate of less than 10% for stage IV patients2,3. Hence, identifying patients with lung cancer at a curable stage is the most effective method to reduce the cancer-related mortality. In the past decades, the radiological features derived from low-dose computed tomography (CT) is the most common non-invasive test for diagnosing lung cancer4,5. However, the clinical application of the low-dose CT is still limited due to the high false positive rate6,7, although it can contribute to a 20% reduction in cancer-related death8.
The plasma cell-free DNA (cfDNA) analysis has offered a promising liquid biopsy biomarker for cancer detection9,10,11. For instance, epigenomics modification of cfDNA, including 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) modifications, are found to manifest cancer pathobiology12,13,14,15,16,17, and have been deployed in lung cancer predictive models18,19,20. Meanwhile, the fragmentomic profiles of cfDNA represents another class of biomarkers for liquid biopsy in oncology and holds promise in cancer detection21. Recent study has revealed the diagnostic potential of fragmentomic features of plasma cfDNA, and further demonstrated the superior performance of 6-mer (6 bp) breakpoint motif over 4 bp end motif in identifying lung adenocarcinoma22.
Nevertheless, the clinical application of the cfDNA analysis would be limited by the potential risk of overfitting accounting for the small sample size, and the diagnostic value of fragmentomic profile from plasma cfDNA 5mC and 5hmC modifications data for lung cancer is still underexplored. Hence, as shown in Fig. 1a, we prospectively enrolled 2032 participants harboring indeterminate pulmonary nodules from different 5 clinical centers, and collected peripheral blood sample before surgery or biopsy to develop an epigenomic dataset. In addition, the diagnostic value of CT-based radiomics features for lung cancer have been extensively proven in previous studies23,24,25,26. To further evaluate the multiomics signature in diagnosing lung cancer, we simultaneously collected the chest CT images from each participant, and constructed a multiomics dataset consisting of epigenomics features and paired radiomics.

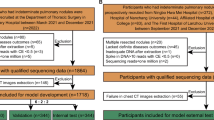
The schematic view for the multiomics dataset. (a) The construction workflow for the multiomics dataset; (b) pathological distribution; (c) information included in this dataset. EDM, end motif; GLRLM, gray-level run length matrix; GLCM, gray-level co-occurrence matrix; NGTDM, neighboring gray tone difference matrix; GLDM, gray-level dependence matrix; GLSZM, gray-level size zone matrix.
Based on the multiomics dataset, we established a multiomics model by integrating clinical, fragmentomic with radiomic features via artificial intelligence technology, namely clinic-RadmC, and demonstrated that clinic-RadmC outperformed single-omics models and clinical model in predicting the malignancy risks of indeterminates pulmonary nodules, offering a more accurate, effective and noninvasive method for diagnosing lung cancer and facilitating individual management27. Therefore, this multiomics dataset would provide multi-dimensional insights for lung cancer detection, and serves as a rich resource contributing to the advancement of lung cancer research.
Methods
Ethical approval
This study was carried out in accordance with the Declaration of Helsinki, and the institutional review boards of all involved centers approved this study (L21-022, L21-022-1, L21-022-2). The recruitment notices for participants in this project was released by the health management department in the media, and all the participants registered at the department of thoracic surgery when they hospitalized. The informed consent form informs participants that their blood samples, anonymized CT image and related clinical data may be used for scientific research and publication of papers. Only by reading the informed consent form and signing the agreement can be proceed to the next step of the process, and all participants signed the informed consent documents. This study also registered in the Chinese Clinical Trial Registry (ChiCTR2100041971), and approved by the Ministry of Science and Technology of China (2021SLCJ2376).
Participants selection
Participants who harbored indeterminate pulmonary nodules were recruited between March 2021 and December 2021 from 5 different centers, including Shanghai Pulmonary Hospital, Ningbo No.2 Hospital, The First Affiliated Hospital of Nanchang University, the Affiliated Hospital of Zunyi Medical College, and The First Hospital of Lanzhou University. The inclusion criteria were: (1) age ≥ 18 years old; (2) without malignant history within 5 years prior to diagnosis; (3) without definite signs of tumor metastasis; (4) did not receive any anti-cancer treatment before diagnosis; and (5) signed an informed consent form. Participants who had multiply resected nodules, had no histopathological outcomes, had inadequate DNA for sequencing after extraction, and showed failure during the sequencing quality control process (spike in DNA > 10 reads with capture efficiency < 0.5, and sequencing reads < one million) were excluded. Further, participants who had no thin-section CT images were also excluded during the dataset processing. The disease outcomes of all participants were surgery-proven, or biopsy-proven. Finally, the dataset was consisted of 1598 (78.6%) lung cancer cases and 434 (21.4%) benign diseases cases. Among them, 1718 participants were enrolled from Shanghai Pulmonary Hospital, and the external test set included 314 participants. The schematic view was provided in Fig. 1b.
Plasma sample collection, cfDNA extraction and sequencing
We collected approximately 10 ml peripheral venous blood samples within 2 days before initial diagnosis from each participant, and performed the plasma isolation. Blood was firstly rested at 4 °C for 30 min, centrifuged at 1000 × g for 10 min at 4 °C, and the supernatant was further centrifuged at 2000 × g for 5 min at 4 °C. Plasma was stored at -80 °C until DNA extraction. cfDNA was isolated using the VAHTS Serum/Plasma Circulating DNA Kit (Vazyme, N902-02) and quantified with a Qubit fluorometer. Libraries were constructed using the VAHTS Universal DNA Library Prep Kit for Illumina (Vazyme, ND607), with spike-in controls (0.01 pg of each amplicon per 10 ng cfDNA). Spike-in controls included three ~180 bp lambda DNA amplicons (unmodified, 5mC-modified, and 5hmC-modified), prepared by PCR amplification (Taq DNA Polymerase, NEB) using dCTP, dmCTP, or 10% dhmCTP/90% dCTP, and purified with AMPure XP beads. Primers sequences were as follows: dCTP FW-5′-CGTTTCCGTTCTTCTTCGTC-3′, RV-5′-TACTCGCACCGAAAATGTCA-3′; dmCTP FW-5′-GTGGCGGGTTATGATGAACT-3′, RV-5′-CATAAAATGCGGGGATTCAC-3′; 10% dhmCTP/90% dCTP FW-5′-TGAAAACGAAAGGGGATACG-3′, RV-5′-GTCCAGCTGGGAGTCGATAC-3′. After that, the 5mC and 5hmC cfDNA sequencing were performed based on the published cfMeDIPseq protocol28,29 and the selective chemical labeling (hMe-Seal)30, which has been detailed described in our recent study27.
Sequencing data processing
The 5mC- and 5hmC-captured libraries were sequenced (150 bp paired-end, 300 cycles) on the Illumina NovaSeq 6000. Low-quality sequences were filtered using Fastp (v0.21.0). Reads were aligned to hg19 and spike-in DNA using BWA (v0.7.17-r1188) with default parameters. SAMtools (v1.7) filtered SAM files (‘-f 3 -F 3852 -q 30’) to retain high-quality, properly paired reads, which were converted to BAM format. Picard (v2.25.5) sorted, indexed, and removed duplicate reads. Bedtools (v2.26.0) converted BAM to BED using ‘-bedpe’, retaining paired-end reads aligned to the same autosome with correct orientation and insert sizes of 20–1000 bp. Spike-in DNA sequences were included in the reference, and capture efficiency (quality control for 5mC/5hmC) was calculated as reads aligned to type-specific spike-in DNA divided by reads aligned to total spike-in DNA.
cfDNA fragmentomic identification
The plasma cfDNA 4 bp EDM were firstly identified using the first 4-nucleotide sequence on each 5′ fragment end of plasma cfDNA after alignment to the reference hg19 genome21. Then, the 4 bp EDM was further extended to 6 bp EDM and identified the first 6-nucleotide sequence on each 5′ end of plasma DNA fragment21. The frequency of each plasma cfDNA end motif was calculated for each sample, and the sum of all EDM frequencies was equal to 1.
Chest CT image collection and scanning parameters
The CT image of participants closest to the date of initial diagnosis were derived from the picture archiving and communication (PACS) system of all involved centers, and The CT scanning parameters of all involved centers were described as follow: (1) Shanghai Pulmonary Hospital: The chest images were acquired using one of the two multi-detector row CT system as follow: (a) Somatom Definition AS (Siemens Medical Systems, Germany) with tube energy as 120 kVp, tube current as 130 mA, rotation time as 0.5 s, detector collimation as 64 × 0.625 mm; and (b) Brilliance 40 (Philips Medical Systems, Netherlands) with tube energy as 120 kV, tube current as 200 mA, rotation time as 0.75 s, detector collimation as 32 × 1.25 mm. All images were reconstructed at 1.0 mm slice thickness and 0.7 mm increment, with the standard soft kernel (Siemens B31 filter, Medical Systems, Germany) and another sharp reconstruction kernel (C filter, Philips Medical Systems, Netherlands); (2) Ningbo No.2 Hospital: All patients underwent routine CT before surgery with a 16-slice spiral CT scanner (Sensation 16; SIEMENS) or a 64-slice spiral CT scanner (SOMATOM Definition; SIEMENS) with the following parameters: 120 kV; 150–200 mA s; pitch, 1; rotation time, 1.0 s; detector collimation, 16 × 0.75 mm or 64 × 0.6 mm; field of view, 500 × 500 mm; matrix, 512 × 512; and slice thickness, 5.0 mm. All images were reconstructed with 3 mm slice thicknesses and 3 mm intervals, and retrieved from the picture archiving and communication system (eWorld, China); (3) Affiliated Hospital of Zunyi Medical College: CT images were acquired by Siemens’ twice generation dual source CT (Somatom Definition Flash, Siemens Healthcare, Forchheim, Germany) and GE Optima CT680 (GE Medical, USA). The Siemens CT scan introduced the optimal automatic adjustment technology of CARE kV tube voltage and CARE Dose4D tube current with reference tube voltages being 100 kV and reference tube current is 350 mAs. The GE CT Tube current modulation technique with a reference tube voltage of 120 kV and a reference tube current of 180 mAs. the pitch is 0.5 and 1.375, and the rotation time is 0.5 s and 0.8 s in the Siemens and GE CT scan, respectively. The remaining scanning parameters are the same: FOV 350 mm × 350 mm (FOV value varies with body type, range is 300mm-350mm); matrix 512 × 512; collimator width 128 × 0.6 mm; reconstruction layer thickness 5 mm; (4) The First Affiliated Hospital of Nanchang University: CT images were obtained from the dual-source CT (Siemens, Germany) with the following scanning parameters: tube voltage = 120 kV, tube current = 150 mAs, scanning layer thickness and layer spacing = 5 mm, and reconstruction layer thickness = 1 mm (1.25 mm); (5) The First Hospital of Lanzhou University: CT images were obtained from 3 helical CT scanners (Discovery CT750 HD, GE Healthcare; Philips iCT 256, Koninklijke Philips N.V.; Somatom Sensation 64, Siemens Healthineers). CT scanning parameters were as follows: (1) tube current adjusted automatically for the Somatom Sensation 64 scanner, (2) tube current 150 to 200 mA for the other two scanners, and (3) CT kernels: Lung kernel for Discovery CT750 HD scanner, YA kernel for Philips iCT 256 scanner, and B60f kernel for Somatom Sensation 64 scanner. For all scanners, tube voltage 120 kVp; tube rotation time of 0.5–1.0 s; field of view (FOV) of 350 mm × 350 mm; matrix, 512 × 512; the layer thickness and spacing were both 5 mm; the reconstruction layer thickness and spacing were both 1.25 mm.
Nodules annotation and segmentation
The private information of participants in primary CT images, such as name, sex, age, birth date, patient ID, institutional information, were firstly deleted via PyDicom package in Python (version 3.7). Then, we converted the anonymized CT data from DICOM format to NIfTI format using the dcm2nii software, and imported it into 3D-Slicer software (version 4.10.1, Brigham and Women’s Hospital) for annotation. The three-dimensional region of interest (3D-ROI) was independently annotated with a bounding box including the primary nodules by two junior thoracic surgeon (M.M. Zhao and J.J. Deng) and a junior radiologist (T.T. Wang) at the lung (level, -450 Hounsfield Unit [HU]; width, 1500 HU) and mediastinal (level, 40 HU; width, 400 HU) window settings. Meanwhile, the 3D-ROIs of 2032 indeterminate pulmonary nodules, which covering the gross volume, were also manually segmented using the 3D-Slicer software (version 4.10.1, Brigham and Women’s Hospital) at the lung (level, -450 Hounsfield Unit [HU]; width, 1500 HU) and mediastinal (level, 40 HU; width, 400 HU) window settings. In cases of disagreement, consensus was achieved by group discussion with a senior thoracic radiologist (X.W. Sun), who provided the majority opinion. All reviewers were blinded to the clinical information during the annotation process.
Classical radiomics feature extraction
The radiomics features were extracted from 3D-ROI using PyRadiomics package in Python (version 3.7), a software adhering to the Image Biomarker Standardization Initiative (IBSI) protocol31, and the steps were performed as recommended by van Timmeren et al.32. The IBSI protocol standardizes the calculation of radiomics features, including first-order statistics, shape-based features, texture features and higher-order feature, and provides definitions and formulas for each feature. Accordingly, 1316 classic radiomics features were extracted, including 252 first-order features, 14 shape-based features, and 1050 texture features (consisting of 336 gray-level co-occurrence matrix [GLCM] features, 224 gray-level size zone matrix [GLSZM] features, 224 gray-level run-length matrix [GLRLM] features, 70 neighboring gray tone difference matrix [NGTDM] features, and 196 gray-level dependence matrix [GLDM] features). All the above procedures were performed in two NVIDIA RTX TITAN GPUs.
Data Records
The information included in the multiomics datasets were illustrated in Fig. 1c. The raw sequencing data have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences33, which could be accessible under the accession number of HRA003308 and HRA007190. The complete CT image dataset has been deposited at Zenodo34,35,36. Additionally, we provided the extracted radiomics features and plasma cfDNA end motif profiles at Figshare37, as well as the clinical data.
For raw sequencing data, each participant has 5mC- and 5hmC-sequencing files. The 5mC-sequencing data are named as “lung-MN-ID.bam”, here “N” represents sequencing batch, and “ID” represents the anonymous code for the participants. The 5hmC-sequencing data are named as “lung-ID.bam”, here “ID” represents the anonymous code for the participants. All the CT images were saved as NIfTI files, and named as “SAMPLE_N.nii”. Here N represents the anonymous code for the participants. Then, the complete CT dataset have been divided and packaged as “Part-I-N.zip” or “supplied(_N).zip”, which are deposited at Zenodo35,36. The annotation and segmentation for each 3D-ROI are exported from 3D-slicer as “R.acsv” and “Segmentation.seg.nrrd”, and saved in a folder, which are named as the corresponding CT image. The complete dataset of annotation and segmentation are then packaged into a “Label.rar” RAR file, and deposited at Zenodo34.
Each participant corresponds to a row in the “clinical_data.csv” file, as detailed in Table 1. For each participant, we collected the sex and age information at the time of initial diagnosis, which we do not share for anonymization reasons. The “Label” column indicated the histopathological status of participants, denoted by a value of 0 (benign) or 1 (cancer). The end motif profiles of 5mC-cfDNA are summarized in “5mC_EDM.xlsx” file, consisting of two spreadsheets. The sheets named as 6 bp represents the information of 6 bp end motifs features from 5mC-cfDNA, and the row includes the SAMPLE_N and 4096 end motifs. The sheets named as 4 bp represents the information of 4 bp end motifs features from 5mC-cfDNA, and the row includes the SAMPLE_N and 256 end motifs. Here N represents the anonymous code for the participants. Similarly, the end motif profiles of 5hmC-cfDNA are summarized in “5hmC_EDM.xlsx” file, consisting of two sheets. The sheets named as 6 bp represents the information of 6 bp end motifs features from 5hmC-cfDNA, and the row includes the SAMPLE_N and 4096 end motifs. The sheets named as 4 bp represents the information of 4 bp end motifs features from 5hmC-cfDNA, and the row includes the SAMPLE_N and 256 end motifs. Here N represents the anonymous code for the participants. The classic radiomics features extracted from 3D-ROI via PyRadiomics package are summarized in “classic_radiomics.csv” file, and the row includes the SAMPLE_N and 1316 radiomics features, while N represents the anonymous code. Furthermore, to match the blood sample to CT images, we provided an additional “ID_matched.csv” file to match patient anonymity codes of sequencing data and CT images. As described in Table 2, “SAMPLE_N” represents the anonymous code of participants providing CT images, “5mC_ID” represents the anonymous code of participants providing 5mC-sequencing data, and “5hmC_ID” represents the anonymous code of participants providing 5hmC-sequencing data. The clinical and multiomics information of participants have been packaged into a “multiomics_data.rar” RAR file, and deposited at Figshare37.
Technical Validation
A unified data quality control and analysis process were used for all the collected peripheral blood sample to ensure that different samples could be compared. During the dataset construction, blood samples from 2459 participants were initially collected, and 100 participants who harboring multiple resected nodules, 110 participants having no definite diseases outcomes were firstly excluded. The blood samples from the rest 2249 samples were used for generating the epigenomics features, and 6 samples were found to have no enough cfDNA extracted for library construction. Then, the capture efficiency of spike in DNA > 10 reads with capture efficiency < 0.5 and the low-quality reads with sequencing reads < one million were adopted as the exclusion criteria, resulting in the exclusion of blood samples from 10 participants. The blood sample operating team was blinded to group status of all samples during the whole process, and only high-quality samples that meet all filtering criteria were included in the multiomics dataset for the subsequent analysis. The t-Distributed Stochastic Neighbor Embedding (t-SNE) analysis revealed that no systematic bias or obvious batch effect for both 5mC- (Fig. 2a) and 5hmC-sequencing data (Fig. 2b) were detected. As shown in Fig. 3a-b, both 5mC- and 5hmC-sequencing data revealed highly specific enrichment. Moreover, the median mapped fragments for the 5mC and 5hmC samples were 12.7 million and 14.1 million, indicating enough fragments for further analyses (Fig. 3c-d). The CT annotation and segmentation process involved a quality control process with two junior thoracic surgeons with over 5 years of experience, one junior radiologist with over 5 years of experience and one senior radiologist with over 30 years of experience. The process followed a workflow where images were firstly randomly assigned to any junior thoracic surgeons or radiologist for initial annotation and segmentation, and then cross-reviewed by each other. If disagreement, the annotation or segmentation would undergo a final check by the senior radiologist to ensure accurate labeling of each CT image.

The t-Distributed Stochastic Neighbor Embedding analysis based on the 6-mer end motifs profiles obtained from the sequencing data. (a), 5mC-sequencing data analysis; (b), 5hmC-sequencing data analysis.

5mC and 5hmC data statistics. Percentage of 5mC (a) and 5hmC (b) reads mapped to the spike-in DNA in the sequencing libraries. The 5mC spike-in DNA is specifically enriched in the 5mC libraries (n = 797), while the 5hmC spike-in DNA is specifically enriched in the 5hmC libraries (n = 1706). Only samples with spike in DNA > 10 reads were included in the enrichment efficiency analysis; Histogram plot of the number of high-quality fragments (paired reads) in 5mC (c) and 5hmC (d) samples (n = 2032).
Usage Notes
To download the raw sequencing data as BAM format, researchers need to apply on the GSA-Human platform at https://ngdc.cncb.ac.cn/gsa-human, and sign the data access agreement. Researchers are requested to register an account at the platform, and then log in to find the sequencing data by entering the access number in the search bar. After that, by clicking the “Request” button and following the steps to make their data access request, the data access agreement could be downloaded during the intermediate process. If approved, researcher would receive an e-mail notification from GSA. Then, they can log in to their account, and select “My requests” to check the status of their application. By clicking on “download” in GSA, they could access the data download address. The end motif profile extracted from the sequencing data could be directly downloaded from Figshare37, and the complete CT image dataset could be directly available from Zenodo35,36, as well as the files of labeling34. The data presented in this manuscript is part of our previously published study27.
Code availability
No custom code was used for constructing this multiomics dataset. The code for end motif extraction could be available at GitHub with link of https://github.com/gangxuehzau/mission/tree/main. The codes for extracting radiomics features was derived from the open-source PyRadiomics package from Python (version 3.7, https://www.python.org).
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA Cancer J Clin 70, 7–30, https://doi.org/10.3322/caac.21590 (2020).
Han, B. et al. Cancer incidence and mortality in China, 2022. Journal of the National Cancer Center https://doi.org/10.1016/j.jncc.2024.01.006 (2024).
Zhang, Y., Luo, G., Etxeberria, J. & Hao, Y. Global Patterns and Trends in Lung Cancer Incidence: A Population-Based Study. Journal of Thoracic Oncology 16, 933–944, https://doi.org/10.1016/j.jtho.2021.01.1626 (2021).
Mazzone, P. J. & Lam, L. Evaluating the Patient With a Pulmonary Nodule. JAMA 327, https://doi.org/10.1001/jama.2021.24287 (2022).
Blandin Knight, S. et al. Progress and prospects of early detection in lung cancer. Open Biology 7, https://doi.org/10.1098/rsob.170070 (2017).
McWilliams, A. et al. Probability of cancer in pulmonary nodules detected on first screening CT. N Engl J Med 369, 910–919, https://doi.org/10.1056/NEJMoa1214726 (2013).
Rob, J. et al. Management of Lung Nodules Detected by Volume CT Scanning. N Engl J Med 361, 2221–2229 (2009).
Aberle, DeniseR. et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 365, 395–409 (2011).
Li, W. et al. Liquid biopsy in lung cancer: significance in diagnostics, prediction, and treatment monitoring. Molecular Cancer 21, https://doi.org/10.1186/s12943-022-01505-z (2022).
Chen, K. et al. Development of new techniques and clinical applications of liquid biopsy in lung cancer management. Science Bulletin 69, 1556–1568, https://doi.org/10.1016/j.scib.2024.03.062 (2024).
Wang, S. et al. Multidimensional Cell-Free DNA Fragmentomic Assay for Detection of Early-Stage Lung Cancer. Am J Respir Crit Care Med 207, 1203–1213, https://doi.org/10.1164/rccm.202109-2019OC (2023).
Hu, X. et al. Integrated 5-hydroxymethylcytosine and fragmentation signatures as enhanced biomarkers in lung cancer. Clinical Epigenetics 14, https://doi.org/10.1186/s13148-022-01233-7 (2022).
Liu, M. C. et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Annals of Oncology 31, 745–759, https://doi.org/10.1016/j.annonc.2020.02.011 (2020).
Liang, W. et al. Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics 9, 2056–2070, https://doi.org/10.7150/thno.28119 (2019).
Rompala, G. et al. Profiling neuronal methylome and hydroxymethylome of opioid use disorder in the human orbitofrontal cortex. Nature Communications 14, https://doi.org/10.1038/s41467-023-40285-y (2023).
Chemi, F. et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nature Cancer 3, 1260–1270, https://doi.org/10.1038/s43018-022-00415-9 (2022).
Vasanthakumar, A. & Godley, L. A. 5-hydroxymethylcytosine in cancer: significance in diagnosis and therapy. Cancer Genetics 208, 167–177, https://doi.org/10.1016/j.cancergen.2015.02.009 (2015).
He, J. et al. Accurate classification of pulmonary nodules by a combined model of clinical, imaging, and cell-free DNA methylation biomarkers: a model development and external validation study. The Lancet Digital Health 5, e647–e656, https://doi.org/10.1016/s2589-7500(23)00125-5 (2023).
Ren, Y. et al. A Highly Sensitive and Specific Non‐Invasive Test through Genome‐Wide 5‐Hydroxymethylation Mapping for Early Detection of Lung Cancer. Small Methods, https://doi.org/10.1002/smtd.202300747 (2023).
Liang, W. et al. Accurate diagnosis of pulmonary nodules using a noninvasive DNA methylation test. Journal of Clinical Investigation 131, https://doi.org/10.1172/jci145973 (2021).
Jiang, P. et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discovery 10, 664–673, https://doi.org/10.1158/2159-8290.Cd-19-0622 (2020).
Guo, W. et al. Sensitive detection of stage I lung adenocarcinoma using plasma cell-free DNA breakpoint motif profiling. EBioMedicine 81, https://doi.org/10.1016/j.ebiom.2022.104131 (2022).
Liu, A. et al. Preoperative diagnosis of malignant pulmonary nodules in lung cancer screening with a radiomics nomogram. Cancer Communications 40, 16–24, https://doi.org/10.1002/cac2.12002 (2020).
Hunter, B. et al. A radiomics-based decision support tool improves lung cancer diagnosis in combination with the Herder score in large lung nodules. EBioMedicine 86, https://doi.org/10.1016/j.ebiom.2022.104344 (2022).
Sun, Y. et al. Radiomics for lung adenocarcinoma manifesting as pure ground-glass nodules: invasive prediction. European Radiology 30, 3650–3659, https://doi.org/10.1007/s00330-020-06776-y (2020).
Massion, P. P. et al. Assessing the Accuracy of a Deep Learning Method to Risk Stratify Indeterminate Pulmonary Nodules. American Journal of Respiratory and Critical Care Medicine 202, 241–249, https://doi.org/10.1164/rccm.201903-0505OC (2020).
Zhao, M. et al. Integrated multiomics signatures to optimize the accurate diagnosis of lung cancer. Nature Communications 16, https://doi.org/10.1038/s41467-024-55594-z (2025).
Shen, S. Y. et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583, https://doi.org/10.1038/s41586-018-0703-0 (2018).
Shen, S. Y., Burgener, J. M., Bratman, S. V. & De Carvalho, D. D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nature Protocols 14, 2749–2780, https://doi.org/10.1038/s41596-019-0202-2 (2019).
Song, C.-X. et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Research 27, 1231–1242, https://doi.org/10.1038/cr.2017.106 (2017).
Zwanenburg, A. et al. The Image Biomarker Standardization Initiative: Standardized Quantitative Radiomics for High-Throughput Image-based Phenotyping. Radiology 295, 328–338, https://doi.org/10.1148/radiol.2020191145 (2020).
van Timmeren, J. E., Cester, D., Tanadini-Lang, S., Alkadhi, H. & Baessler, B. Radiomics in medical imaging—“how-to” guide and critical reflection. Insights into Imaging 11, https://doi.org/10.1186/s13244-020-00887-2 (2020).
Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2024. Nucleic Acids Res 52, D18-d32, https://doi.org/10.1093/nar/gkad1078 (2024).
Zhao, M. & She, Y. CT labeling for “A multiomics dataset of paired CT image and plasma cell-free DNA end motif for patients with pulmonary nodules”. Zenodo https://doi.org/10.5281/zenodo.15043680 (2025).
Zhao, M. & She, Y. CT dataset for “Integrated multiomics signatures to optimize the accurate diagnosis of lung cancer” Part-I. Zenodo https://doi.org/10.5281/zenodo.13908014 (2024).
Zhao, M. & She, Y. CT dataset for “Integrated multiomics signatures to optimize the accurate diagnosis of lung cancer” Part 2. Zenodo https://doi.org/10.5281/zenodo.13908029 (2024).
Zhao, M. & She, Y. Multiomics dataset for “A multiomics dataset of paired CT image and plasma cell-free DNA end motif profiles for patients with pulmonary nodules”. figshare. Dataset. https://doi.org/10.6084/m9.figshare.26861077.v2 (2024).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (92474114 to Y.L.S.; 92259205, 91959126 to C.C.; 82241238 to D.X.; 82241237 to L.X.L.; 82302317 to B.X.H), Ningbo Top Medical and Health Research Program (No.2022030208 to G.F.Z.), Clinical Research Foundation of Shanghai Pulmonary Hospital (FKLY20016, Fkcy2410 to M.M.Z.; 2021LY0311 to C.C.), The 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYYC23024 to D.X.), Research Fund of West China Hospital (2020HXFH006 to D.X.) and the China Postdoctoral Science Foundation (2021M700341 to B.X.H). We would like to thank the MultiomIcs claSSIfier for pulmOnary Nodules (MISSION) Collaborative Group for their supports and efforts. We are also grateful to the clinical research coordinators, Xiaomin Zhu and Chong Zhu, who helped to collect the plasma samples. Then, we thank all the participants, without whom this research would not have been possible.
Author information
Authors and Affiliations
Contributions
Conception and design: M.M. Zhao, J.J. Deng, D.P. Zhao, C. Chen, and Y.L. She. Acquisition of data (acquired and managed patients, provided facilities, etc.): M.M. Zhao, T.T. Wang, Y.F. Zhong, S.H. Li, Y. Wang, Y.M. He, T. Chen, L.N. Guo, W.D, Qu, M.L. Yang. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): M.M. Zhao, G. Xue, B.X. He. Writing, review, and/or revision of the manuscript: M.M. Zhao, G. Xue, B.X. He, J.J. Deng, Y.L. She. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Y.X. Song, G.F. Zhao, B.T. Yu, M.J. Ma, X.W. Sun, D.X. Study supervision: D.P. Zhao, C. Chen, and Y.L. She.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, M., Xue, G., He, B. et al. A multiomics dataset of paired CT image and plasma cell-free DNA end motif for patients with pulmonary nodules. Sci Data 12, 545 (2025). https://doi.org/10.1038/s41597-025-04912-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04912-1
