
Abstract
The whitespotted conger (Conger myriaster) is an ecologically and economically significant benthic marine species widely distributed across East Asia’s coastal waters. Despite this importance, the genomic resources for this species remain limited, hindering evolutionary and aquaculture research. Here, we present the first high-quality chromosome-level genome assembly of C. myriaster using PacBio CLR, WGS, 10X Genomics and Hi-C data. The resulting 1.09 Gb genome assembly exhibits excellent contiguity, with 97.49% of sequences anchored onto 19 chromosomes. The assembled genome achieved a BUSCO completeness stands at 98.00%, containing 34.80% repetitive sequences and 24,063 predicted protein-coding genes. This foundational genomic resource overcomes a major limitation, providing the essential framework for future investigations into the evolutionary adaptations and for the genetic improvement of C. myriaster in aquaculture.
Similar content being viewed by others


Background & Summary
The whitespotted conger (Conger myriaster) is a member of the order Anguilliformes, family Congridae, and widely distributed in the East China Sea, Yellow Sea, and the Bohai Bay in China, and from southern Hokkaido to northern Okinawa in Japan, and around the Korean Peninsula1. Despite its ecological and economic importance, knowledge of its life history is limited. Furthermore, exemplified by its complex leptocephalus larval stage, which exhibits unique ecological traits distinct from juveniles and adults and utilizes major ocean currents, such as the North Equatorial Current and the Kuroshio Current, for dispersal—a strategy also observed in eels like Anguilla japonica and Anguilla marmorata1,2,3, and successful artificial breeding has yet to be achieved4. At present, C. myriaster remains entirely reliant on wild catches for consumption, with limited genomic resources available to support conservation and breeding efforts5.
Indeed, C. myriaster faces significant threats from overfishing and marine pollution, leading to a steady decline in its population6, and the urgency for improved management and conservation measures in primary fishing grounds has become increasingly evident, driven by ecological pressures and the imperative to ensure sustainable fisheries7. While the Yellow Sea Fisheries Research Institute has made progress in artificial breeding and aquaculture research8,9, immature individuals from offshore fisheries present significant challenges for further studies. Critically, underlying many of these challenges is the lack of published genomic resources for C. myriaster.
In this study, we present the first high-quality chromosome-level genome assembly of C. myriaster using an integrated strategy which combines Whole Genome Sequencing (WGS), 10X Genomics, PacBio Continuous Long Reads (CLR), and Hi-C data. The assembled genome has a total size of 1.09 Gb, with 97.49% of the sequences anchored to the 19 chromosomes as previously identified through karyotype analysis10. The assembly achieved N50 lengths of 16.76 Mb for contigs and 58.40 Mb for scaffolds, and 34.80% of the genome was annotated as repetitive sequences. A total of 24,063 protein-coding genes were predicted, with 99.80% of them functionally annotated. This high-quality genome assembly provides a robust foundation for developing molecular markers and advancing conservation and aquaculture efforts for C. myriaster. Furthermore, it serves as a critical resource for investigating the evolutionary dynamics and phylogenetics of eel species.
Methods
Sample collection and sequencing
The present study was dedicated to the genome sequencing of an adult female C. myriaster. The specimen was sourced from a local aquaculture facility (Haiyang Yellow Sea Fisheries Co., Ltd.), with stringent selection criteria to ensure optimal health status. Prior to sampling, the fish was subjected to a mild anesthetic protocol to alleviate stress and ensure humane handling.
For CLR data, sequencing was performed on the PacBio Sequel II platform with blood tissue using the CLR mode. This mode is particularly advantageous for obtaining long reads, which are essential for accurate genome assembly and resolving complex genomic regions. The sequencing run generated 104.76 Gb of raw data, providing approximately 95-fold depth of the fish genome. This high depth ensures robust representation of the genome and facilitates the detection of low-frequency variants and structural variations (Table 1).
For Hi-C data, the library was prepared by first crosslinking cells with formaldehyde. DNA was then digested using the MboI restriction enzyme, followed by end-filling and biotin labeling. The resulting blunt-end fragments were ligated, purified, and sheared into 300–500 bp fragments. Quality control was conducted using Qubit 2.0, an Agilent 2100 instrument (Agilent Technologies, CA, USA), and q-PCR. Finally, 150 bp paired-end sequencing was performed on the Illumina platform with blood tissue, yielding 117.70 Gb of Hi-C data, which provided approximately 107-fold depth of the fish genome (Table 1).
For WGS data, genomic DNA extracted from muscle tissues was fragmented to approximately 350 bp using E220 Covaris (Covaris Inc., USA). The fragmented DNA underwent 3′ end-repair, adaptor ligation, and amplification via ligation-mediated polymerase chain reaction (LM-PCR). Single-stranded DNA molecules were separated and circularized, followed by rolling-circle amplification (RCA) to generate DNA nanoballs (DNBs). These DNBs were loaded onto patterned nanoarrays of the BGI-Seq 500 platform and sequenced using PE100 + 10 chemistry, yielding 100 bp paired-end reads with 10 bp dual-index barcodes for sample demultiplexing. A total of 39.46 Gb of short-read data was generated, achieving approximately 36-fold depth of the fish genome (Table 1).
For 10X Genomics data, DNA was processed using the 10X Genomics Chromium platform to generate a library with long-range genomic information. The library was then sequenced on the BGI-Seq 500 platform with muscle tissue, leveraging its high-throughput capabilities and advanced sequencing chemistry. This combination yielded a total of 203.73 Gb of data, achieving approximately 186-fold depth of the fish genome (Table 1).
For full-length transcript sequencing, RNA was extracted from a mixed tissue sample (eye, intestine, spleen, kidney, testis, ovary, pituitary, liver, muscle, brain, skin, gill, heart, and stomach), and qualified RNA samples underwent reverse transcription, end repair, DNA fragmentation, adapter ligation, and amplification to construct the library. Sequencing was performed on the PacBio Sequel IIe platform, yielding 32.76 Gb of Circular Consensus Sequencing (CCS) data (Table 1), thereby facilitating precise genome annotation by resolving complex gene structures.
Genome estimate and assembly
The estimated sizes of the predicted genomes were determined using 10X Genomics reads alongside JELLYFISH11 (v2.2.3) with a k-mer size of 19-mer frequency depth distribution and subsequently analyzed with GenomeScope12 (http://genomescope.org/) to generate the k-mer frequency distribution plot. K-mer analysis suggests that the genome spans approximately 936.65 Mb, with 259.55 Mb consisting of repeated sequences and a heterozygosity rate estimated at 1.04% (Fig. 1). To assemble a high-quality genome for the whitespotted conger, we first generated a draft genome using CLR data with NextDenovo13 (v2.5.2). The draft genome was then polished with error-corrected CLR, 10X Genomics and WGS data using NextPolish14 (v1.4.1). Scaffolding was performed with yahs15 (v1.2.2) in combination with Hi-C data, resulting in 19 chromosomes significantly longer than the other scaffolds, consistent with the chromosome number reported in prior karyotype analyses10 (Fig. 2a,b and Table 2). Final manual refinement was completed with Juicer-box16 (v1.91), producing a 1.09 Gb genome (Fig. 3a and Table 3). Utilizing MCScanX17 and SynVisio18 to generate synteny plots reveals significant genomic synteny between the genomes of C. myriaster and its congeneric species Conger conger19, indicating that the assembled chromosomes possess reliability (Fig. 3b).This genome size aligns with the C-value estimated in the ANIMAL GENOME SIZE DATABASE (https://www.genomesize.com), with scaffold N50 of 58.40 Mb (Fig. 2a).

Genome size estimates for C. myriaster using Kmer-based method.

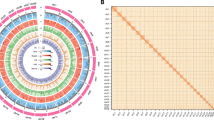
C. myriaster genome snail plot and circos plot. (a) The BlobToolKit snail plot displays the scaffold N50 metric and BUSCO gene completeness. The main plot is divided into 1,000 size-ordered bins, with each bin representing 0.10% of the 1,093,778,583 bp assembly. The gray area shows the distribution of sequence lengths, and the radius of the plot is scaled to the longest sequence in the assembly (91,834,031 bp, highlighted in red). The orange and light orange arcs represent the scaffold N50 and N90 sequence lengths (58,402,862 bp and 39,753,706 bp, respectively). The light gray spiral displays the cumulative sequence count on a logarithmic scale, with white tick marks indicating successive orders of magnitude. The outer blue and light blue regions illustrate the distribution of GC, AT, and N percentages within the same bins as the inner plot. In the upper right corner, a summary of BUSCO genes from the actinopterygii_odb10 dataset is shown, including complete, fragmented, duplicated, and missing BUSCOs. (b) The Circos plot illustrates the GC content, gene density, repetitive sequences, and collinearity between chromosomes in the assembled genome of C. myriaster. The Circos plot represents fundamental information about the genome. The outermost layer displays the chromosome names and their lengths. Moving inward, the purple band represents GC content, the blue band indicates gene density, and the green band denotes repetitive sequences. The innermost part illustrates the synteny among chromosomes within C. myriaster.

Hi-C heatmap and collinearity dot plot of the genome. (a) The Hi-C heatmap illustrates the chromosome interaction frequencies of C. myriaster genome, with each blue contour representing a chromosome. (b) The dot plot displays the collinearity relationships between the genome assembly of C. myriaster and C. conger.
Repetitive sequence annotation
To annotate the repetitive elements in the genome of C. myriaster, we employed a combination of de novo prediction and homology-based annotation methods. For de novo prediction, LTR retrotransposons were predicted using LTR-FINDER-parallel20 (v1.1), while de novo repetitive elements were identified with RepeatModeler21 (v2.0.6). The resulting predictions were integrated into a genome-specific repeat element database of C. myriaster of the repeat sequences. For the homology-based method, RepeatProteinMask and Repbase modules in RepeatMasker22 (v.4.1.7) were used for predicting based on homologous sequences in RepBase database (http://www.girinst.org/repbase) and Dfam database23 (v3.8). In total, 380.59 Mb consisted of repetitive sequences, accounting for 34.80% of the genome assembly (Fig. 2b and Table 4).
Genomic structure and functional annotation
Building upon the masked repetitive sequences of C. myriaster, we employed braker324 (v3.0.8) with default parameters for de novo gene prediction. Furthermore, we leveraged Miniprot25 (v0.13) for homology-based annotation of protein sequences from three species, including C. conger, Anguilla anguilla, and Anguilla rostrata. Concurrently, we aligned RNA-seq data (SRP36125626) from the National Center for Biotechnology Information (NCBI) using HISAT227 (v2.2.1) to the genome and subsequently assembled the transcriptome with StringTie28 (v2.2.3). TransDecoder29 (v5.7.1) was utilized to further identify open reading frames (ORFs) within the assembled transcripts, predicting potential coding regions to construct annotation evidence. We processed full-length transcriptome data using the ISOseq 330 (v4.0.0) pipeline and aligned them to the genome using GMAP31 (2021-08-25) to generate annotation evidence. Finally, we integrated four annotation strategies—de novo, homology-based, transcript-based, and full-length transcriptome—using EvidenceModeler32 (v1.1.1). We also used the annotations from EGAPx33 (v0.3.1-alpha) and RNA-seq data as references, comparing them with C. myriaster annotations to further refine the final gene set, thereby identifying 24,063 protein coding genes.
We used Diamond34 (v2.1.6) to align the predicted gene protein sequences with various functional databases, setting an E-value threshold of 1e-5. These databases included KEGG35, Swiss-Prot36, EggNOG37, Pfam38, Kofam39, and the Non-Redundant40 (NR) database, to extract potential gene functional information for subsequent statistical analysis. A total of 24,016 genes, accounting for 99.80% of the estimated total number of protein-coding genes, were effectively annotated by at least one of these databases (Fig. 4 and Table 5).

Upset plot of gene functional annotation using data from EggNOG, Pfam, KEGG, NR, Kofam, and SwissProt.
Data Records
The WGS (SRR32021150), PacBio CLR (SRR32021152), Hi-C (SRR32021151), 10X Genomics (SRR32021149), RNA (SRR32021148) data used for the genome have been deposited in the NCBI Sequence Read Archive (SRA) under the accession number SRP55777641. And the chromosome-level assembly of C. myriaster was deposited in the National Center for Biotechnology Information (NCBI) under the accession number GCA_047653785.142. The chromosome assembly of C. myriaster and genomic annotation results can be found in the figshare dataset under DOI code: https://doi.org/10.6084/m9.figshare.2812451943.
Technical Validation
To assess the accuracy of gene annotation, we compared the distribution of gene length, exon length, CDS length and intron length in C. myriaster with gene data from C. conger, A. anguilla44, and A. rostrata45.The results indicated a high degree of similarity in gene composition distribution among the three species (Fig. 5). We utilized BUSCO46 (v5.3) with the Actinopterygii database (actinopterygii_odb10) to evaluate the completeness of our genome assembly. The BUSCO analysis indicated an overall completeness of 98.00%, with 91.80% being single-copy, 6.20% being duplicated, 0.70% being fragmented, and 1.30% being missing (Fig. 2a and Table 2). We mapped WGS data to the genome using Merqury47 (v1.3) and minimap248 (v2.28-r1209), achieving a quality score of 32.63 (as indicated by the quality value, QV) and a read-to-contig alignment rate of 99.49%. Additionally, CRAQ49 (v1.0.9-alpha) assesses the quality of genome assembly by aligning raw CLR reads to the assembled genome. The assembly quality for small regions and large structural segments can be calculated as 97.70 (R-AQI) and 100 (S-AQI), respectively, indicating that the genome has achieved reference quality (>90).

Comparisons of gene, exon, CDS, and intron lengths of C. myriaster and the three closely related species (C. conger, A. Anguilla and A. rostrata).
Code availability
In this study, we did not employ any customized scripts or software for personalized analysis. The analytical tools and parameters used are described in the methods section. For software without specific parameter descriptions, the default parameters were selected.
References
Kurogi, H., Mochioka, N., Takaki, Y. & Tabeta, O. First offshore record of Conger myriaster leptocephali in the East China Sea. Fish. Sci. 68, 1155–1157, https://doi.org/10.1046/j.1444-2906.2002.00547.x (2002).
Miller, M. J. et al. Offshore spawning of Conger myriaster in the western North Pacific: evidence for convergent migration strategies of anguilliform eels in the Atlantic and Pacific. Naturwissenschaften 98, 537–543, https://doi.org/10.1007/s00114-011-0787-y (2011).
Kurogi, H. et al. Discovery of a spawning area of the common Japanese conger Conger myriaster along the Kyushu-Palau Ridge in the western North Pacific. Fish. Sci. 78, 525–532, https://doi.org/10.1007/s12562-012-0468-6 (2012).
Chen, Y. et al. Morphometric analysis and influence of important morphological traits on the body weight and net body weight of Conger myriaster in an industrialized culture model. Fish. Sci. Adv. 45, 83–96, https://doi.org/10.19663/j.issn2095-9869.20230816001 (2024).
Wu, M. et al. Changes of individual morphology and oocyte characteristics and analysis of tissue nutrient content during the ovary development of conger eel (Conger myriaster). J. Shanghai Ocean Univ. 30, 442–453, https://doi.org/10.12024/jsou.20200503026 (2021).
Hori, T. et al. Effects of water temperature on white-spotted conger Conger myriaster activity levels determined by accelerometer transmitters. Fish. Sci. 85, 295–302, https://doi.org/10.1007/s12562-019-01289-0 (2019).
Kawazu, M. et al. Biological characteristics of Conger myriaster during the initial stage of spawning migration in the East China Sea. Fish. Sci. 81, 663–671, https://doi.org/10.1007/s12562-015-0893-4 (2015).
Shi, B. et al. A study on sperm ultrastructure in the whitespotted conger, Conger myriaster. Mar. Sci. 47, 112–120, https://doi.org/10.11759/hykx20221215001 (2023).
Zhao, X. et al. Effects of dietary supplementation of compound probiotics on growth, non-specific immunity, intestinal digestive enzyme, and microbiota of whitespotted conger (Conger myriaster) in recirculating aquaculture system. Aquac. Int. 32, 2713–2738, https://doi.org/10.1007/s10499-023-01293-3 (2024).
Ojima, Y. & Ueda, H. A Karyotypical Study of the Conger Eel (Conger myriaster) in in vitro Cells, with Special Regard to the Identification of the Sex Chromosome. Proc. Jpn. Acad. 58, 56–59, https://doi.org/10.2183/pjab.58.56 (1982).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107, https://doi.org/10.1186/s13059-024-03252-4 (2024).
Hu, J. et al. NextPolish: a fast and efficient genome polishing tool for long read assembly. Bioinformatics 35, 5143–5149, https://doi.org/10.1093/bioinformatics/btz891 (2019).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808, https://doi.org/10.1093/bioinformatics/btac808 (2023).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Bandi, V. et al. in Plant Bioinformatics: Methods and Protocols (ed. Edwards, D.). 285–308 (Springer US, 2022).
Adkins, P., et al. The genome sequence of the European conger eel, Conger conger (Linnaeus, 1758). Wellcome Open Res 9, 532, https://doi.org/10.12688/wellcomeopenres.23052.1.
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA 10, 48, https://doi.org/10.1186/s13100-019-0193-0.
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. U.S.A. 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.1–4.10.14, https://doi.org/10.1002/0471250953.bi0410s05 (2009).
Storer, J. et al. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA 12, 2, https://doi.org/10.1186/s13100-020-00230-y (2021).
Gabriel, L., et al. BRAKER3: fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiv 2023.06.10.544449, https://doi.org/10.1101/2023.06.10.544449 (2024).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014, https://doi.org/10.1093/bioinformatics/btad014 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP361256 (2023).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Haas, B. J. et al. TransDecoder v5.7.1. GitHub. https://github.com/TransDecoder/TransDecoder (2024).
Guizard, S. et al. nf-core/isoseq: simple gene and isoform annotation with PacBio Iso-Seq long-read sequencing. Bioinformatics 39, btad150, https://doi.org/10.1093/bioinformatics/btad150 (2023).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875, https://doi.org/10.1093/bioinformatics/bti310 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
EGAPx. GitHub. https://github.com/ncbi/egapx (2024).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368, https://doi.org/10.1038/s41592-021-01101-x (2021).
Kanehisa, M, & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27-30, https://doi.org/10.1093/nar/28.1.27 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48, https://doi.org/10.1093/nar/28.1.45 (2000).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314, https://doi.org/10.1093/nar/gky1085 (2019).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2021).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252, https://doi.org/10.1093/bioinformatics/btz859 (2020).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504, https://doi.org/10.1093/nar/gki025 (2005).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP557776 (2025).
Han, S. et al. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_047653785.1 (2025).
Han, S. Conger myriaster Genomic Dataset: Assembly, Annotation, and Protein Data. figshare https://doi.org/10.6084/m9.figshare.28124519.v2 (2025).
Jansen, H. J. et al. Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci. Rep. 7, 7213, https://doi.org/10.1038/s41598-017-07650-6 (2017).
Pavey, S. A. et al. Draft genome of the American Eel (Anguilla rostrata). Mol. Ecol. Resour. 17, 806–811, https://doi.org/10.1111/1755-0998.12608 (2017).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: novel and streamlined workflows along with broader and deeper phylogenetic depth for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Li, K. et al. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat Commun 14, 6556, https://doi.org/10.1038/s41467-023-42336-w (2023).
Acknowledgements
This research was funded by the National Key R&D Program of China (grant number 2022YFD2400100), the Key Research and Development Project of Shandong Province (2024LZGC005), the AoShan Talents Cultivation Program Supported by Qingdao National Laboratory for Marine Science and Technology (grant number 2017ASTCP-ES06), the Taishan Scholars Program (NO. tstp20221149) to C.S, the National Ten-Thousands Talents Special Support Program to C.S, the Central Public-interest Scientific Institution Basal Research Fund, CAFS (grant number 2023TD19 and 20603022023023), the Key R&D Program of Hebei Province, China (21326307D), the China Agriculture Research System (grant number CARS-47-G03) and the project by the National Marine Genetic Resource Center, the National Key R&D Program of China (grant number 2024YFD2401002).
Author information
Authors and Affiliations
Contributions
C.S. and B.S. conceived of the project. S.H. and C.L. analyzed and drafted the data. S.L., C.L., K.L., H.W., and Q.W. revised the manuscript. Y.C. and C.W. prepared the sample materials and extracted the DNA. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, S., Li, C., Li, S. et al. High-quality chromosome-level genome assembly of the whitespotted conger (Conger myriaster). Sci Data 12, 626 (2025). https://doi.org/10.1038/s41597-025-04947-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04947-4
