
Abstract
Tuberculosis (TB), caused by the Mycobacterium tuberculosis complex (MTBC), remains a major global health challenge. Whole genome sequencing (WGS) offers an invaluable tool for understanding the genetic diversity and drug resistance profiles of MTBC. This study provides a comprehensive WGS dataset of 2,520 MTBC isolates collected from four Italian regions—Lombardy, Piedmont, Emilia-Romagna, and Lazio—between 2017 and 2020. The dataset includes genomic data along with associated metadata, such as geographic location and drug susceptibility profiles, providing a robust resource for studying TB epidemiology and transmission dynamics. This collection represents the largest publicly available MTBC WGS dataset from Italy and has been validated to ensure accuracy and completeness. By making this dataset accessible, we aim to support collaborative research, facilitate the exploration of MTBC evolution and drug resistance, and enhance TB surveillance efforts.
Similar content being viewed by others
Background & Summary
Tuberculosis (TB), caused by members of the Mycobacterium tuberculosis complex (MTBC), remains a significant global health challenge1. In 2023, an estimated 10.8 million people globally contracted tuberculosis (TB), resulting in approximately 1.25 million deaths1. These figures have risen since the onset of the COVID-19 pandemic. TB continues to be a major public health issue, particularly in regions with high HIV prevalence and limited healthcare access1,2. The emergence of drug-resistant strains, along with challenges in diagnosis, treatment, and healthcare delivery, underscores the urgent need for innovative approaches to combat TB. In 2023, there were approximately 400,000 cases of multidrug-resistant tuberculosis (MDR-TB), resulting in about 182,000 deaths globally1. Only about one-third of subjects with MDR-TB had access to appropriate treatment1. Effective surveillance of MTBC lineages and their associated drug resistance patterns is essential for tracking transmission dynamics, identifying high-risk strains, and guiding tailored public health interventions. Certain MTBC lineages, such as Lineage 2 (Beijing), have been associated with a higher propensity for drug resistance and increased transmissibility3, which can impact treatment outcomes and control strategies. Monitoring lineage distribution helps to predict resistance trends, improve diagnostics, and optimize treatment regimens, ultimately enhancing TB control efforts.
Whole genome sequencing (WGS) has emerged as a powerful tool in the fight against TB, enabling comprehensive genetic characterization of MTBC strains and shedding light on their epidemiology, evolution, and drug resistance profiles4,5,6,7. By providing high-resolution data on genomic variations, WGS facilitates the tracking of transmission chains, identification of outbreaks, and detection of resistance mutations with unprecedented accuracy and speed7,8,9.
Italy presents a unique setting for TB epidemiology being situated at the crossroads of Europe10. Although it has long been among the low TB incidence countries, the rise of drug-resistant strains, population mobility, and healthcare inequalities can affect TB epidemiology11,12. In this context, we present a comprehensive dataset comprising the whole genome sequences of MTBC strains collected from diverse geographic regions within Italy13. This dataset represents a valuable resource for researchers and public health authorities, offering insights into the genetic diversity, transmission dynamics, and drug resistance patterns of MTBC strains circulating in the country. By sharing this dataset openly, we aim to foster collaboration, accelerate research efforts, and contribute to the global fight against TB.
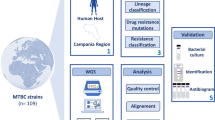
The primary aim of our investigation, conducted as part of the CCM-2018 project (National Center for Disease Prevention and Control), titled ‘Definition of Strategies for Controlling Tuberculosis Associated with HIV in Italy within the Context of a Tuberculosis Elimination Strategy,’ was to construct a comprehensive dataset of WGS data from 2,520 MTBC-positive individuals diagnosed in Piedmont, Lombardy, Emilia-Romagna, and Lazio from January 2017 to June 2020 (Fig. 1)13. The percentage of MTBC isolates analyzed through WGS relative to the total culture-confirmed cases during this period was 34.7% in Piedmont, 38.3% in Emilia-Romagna, 46.4% in Lombardy, and 84.2% in Lazio. This dataset constitutes the largest collection of MTBC isolates from Italy to date, offering critical insights into the genetic diversity, transmission dynamics, and drug resistance patterns of TB strains in the country. WGS analysis was also employed to identify drug resistance profiles (Table 1) and characterize diverse MTBC lineages across the four regions (Fig. 2a) and within different drug resistance profiles (Fig. 2b). This approach enhanced our understanding of regional Mycobacterium tuberculosis variants and strengthened infection surveillance efforts by providing detailed genetic insights.

Workflow Overview for MTBC Genomic and Drug Resistance Profiling. This figure illustrates the workflow used for the genomic and drug resistance profiling of Mycobacterium tuberculosis complex (MTBC) isolates across four Italian regions: Emilia-Romagna, Lazio, Lombardy, and Piedmont. The process begins with data collection (left panel), where MTBC isolates from tuberculosis patients are gathered, and phenotypic drug susceptibility testing (pDST) and HIV status are recorded. Whole-genome sequencing (WGS) is performed on the isolates. In the data analysis stage (middle panel), sequencing data undergo quality control (QC) of FASTQ files, followed by mapping to a reference genome. Variant calling is conducted, generating BAM and VCF files for further analysis. The data generation phase (right panel) includes drug resistance profiling, genotyping, and phylogeny or cluster analysis to study the genetic relationships between isolates. Finally, technical validation (far right panel) is performed to confirm the accuracy of variant calling and resistance profiling through drug susceptibility tests and validation of VCF files. This workflow outlines the integration of WGS data with clinical and technical validation to study MTBC in the context of drug resistance and epidemiology. Figure created using BioRender.com by Arash Ghodousi with license to publish.

(a) Histogram plot showing the percentage distribution of Mycobacterium tuberculosis complex (MTBC) lineages and sublineages across four Italian regions: Emilia-Romagna, Lazio, Lombardy, and Piedmont. The X-axis represents different MTBC lineages, while the Y-axis shows the percentage of isolates for each lineages and sublineages. This figure categorizes the genetic diversity of MTBC isolates into lineages and sublineages as follows: Lineage 1 includes EAI and EAI Manila; Lineage 2 is represented by Beijing; Lineage 3 by Delhi-CAS; Lineage 4 encompasses Haarlem, LAM, Cameroon, X-type, Ural, S-type, mainly-T, Euro-American and H37Rv-like; Lineage 5 and 6 are represented by West-Africa 1 and West-Africa 2, respectively. (b) This figure represents the percentage distribution of MTBC lineages across different drug resistance profiles: Pan-susceptible (Pan-S), Rifampicin-resistant/Multidrug-resistant (RR/MDR), Pre-extensively drug-resistant/Extensively drug-resistant (Pre-XDR/XDR), and Other*. The X-axis shows the drug resistance profiles, while the Y-axis indicates the percentage of isolates for each lineage. Different colors represent the MTBC lineages, with Lineage 4 (Euro-American) being dominant across most drug resistance profiles. The figure highlights the correlation between specific MTBC lineages and their drug resistance profiles. *Note: The “Other” category includes Mono/Poly resistance to other anti-TB drugs not classified within the RR/MDR, or Pre-XDR/XDR categories.
Among the isolates in this dataset13, 94 (3.7%) were identified as rifampicin-resistant (RR-TB), with 83 (88.3%) of these also resistant to isoniazid (i.e., MDR-TB). Additionally, 22 (23.4%) of the MDR-TB strains were resistant to fluoroquinolones, classifying them as pre-XDR-TB (Table 1). The phylogenetic analysis of our dataset revealed that Lineage 4 (Euro-American) was predominant, encompassing 1,859 strains (73.5% of the total), with the sub-lineages Haarlem, T, and LAM representing 555 (29.8%), 505 (27.1%), and 310 (16.7%) strains, respectively. Lineage 3 (Delhi-CAS) accounted for 202 (8.0%) strains, Lineage 2 (Beijing) for 185 (7.4%) strains, and Lineage 1 (EAI) for 166 (6.6%) strains. Additionally, the dataset included 51 M. bovis strains. The less prevalent Lineages 5 (West Africa 1), 6 (West Africa 2), and 7 (Ethiopian) collectively constituted less than 2% of the analyzed strains, as illustrated in Fig. 2a,b13.
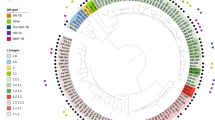
Transmission clusters were analyzed with a 5-SNP threshold, identifying a total of 84 clusters comprising 432 (18%) strains with three or more members (Fig. 3). The largest cluster identified in the study, consisting of 35 MTBC isolates, belonged to the Beijing lineage (Lineage 2). This lineage is known for its global distribution and association with increased virulence and drug resistance14,15.

Maximum Likelihood Phylogenetic Tree of Mycobacterium tuberculosis complex (MTBC) isolates. This figure displays a maximum likelihood phylogenetic tree, constructed from whole genome sequencing data of 2,520 MTBC isolates, gathered from four Italian regions—Piedmont, Lombardy, Emilia-Romagna, and Lazio—between January 2017 and June 2020. Each tip on the tree denotes a unique isolate. Branches are color-coded to illustrate various attributes: sublineages are represented from the innermost part of the tree, followed by broader lineages, types of infection (Pulmonary vs extra-pulmonary), HIV status, clustering status (Group) which is determined by a 5-SNP threshold, and drug resistance profiles. RR: Rifampicin resistant. MDR: Multidrug-resistant. Pre-XDR: Pre-extensively drug-resistant. XDR: Extensively drug-resistant.
Overall, this comprehensive collection of WGS data serves as a robust foundation for investigating the biological impact of MTBC distribution. By offering high-resolution genomic insights, it allows researchers to conduct detailed comparative analyses with other global and regional data, identifying key lineages and pinpointing mutations associated with drug resistance. The dataset is further enriched by the inclusion of detailed clinical and demographic parameters such as patient origin, types of TB infection (pulmonary vs. extrapulmonary TB), initial biological samples, HIV status, and DST results13. These additional factors provide unique opportunities to explore the complex interactions between demographic and clinical characteristics and MTBC infection dynamics. The diverse range of genetic variation and presence of drug-resistant strains within this collection adds significant value to research efforts, allowing for comprehensive investigations into the evolution, transmission, and resistance mechanisms of MTBC. This resource is readily available for immediate use in studies that correlate phenotypic traits with genotypic data, thereby facilitating the identification of drug-resistance mutations and markers crucial for understanding disease progression and developing targeted therapies. Furthermore, the depth and breadth of this WGS collection make it an essential tool for advancing TB research. It offers critical insights necessary for formulating more effective TB management and control strategies, contributing significantly to the global effort to combat tuberculosis, particularly in the context of emerging drug-resistant strains.
Methods
Study population and sample collection
We conducted a retrospective analysis of MTBC strains collected from four regions in Italy: Lombardy, Lazio, Piedmont, and Emilia-Romagna, spanning the period from January 2017 to June 2020. The study population comprised individuals diagnosed with pulmonary and extra-pulmonary TB, including both HIV-positive and HIV-negative individuals. Clinical specimens were obtained from patients presenting with TB symptoms at participating healthcare facilities. Samples were categorized based on TB type (pulmonary vs. extra-pulmonary) and HIV status (positive vs. negative). A total of 2935 MTBC strains were collected, with 2731 subjected to WGS. High-quality sequences were obtained from 2529 strains. To maintain the integrity of the epidemiological data, 9 re-isolates were excluded, leaving 2520 unique clinical isolates for analysis, representing 85.9% of the initially collected strains (Table 2).
DNA extraction
Genomic DNA was extracted from cultured MTBC isolates using the Maxwell 16 Instrument (Promega Corporation, Madison, WI, USA) or the QIAamp DNA minikit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Isolates grown on solid media (Löwenstein-Jensen agar) had bacterial colonies aseptically transferred to microcentrifuge tubes, while those in liquid media (MGIT 960 or Middlebrook 7H9 Broth) were processed by centrifuging aliquots to pellet the cells. The extraction, performed with the Maxwell 16 Tissue DNA Purification Kit, involved lysing cells to release DNA, which was then bound to purification cartridges. Subsequent steps included washing to remove contaminants and eluting purified DNA in a final volume for downstream applications. The DNA samples used in this study are available at the TB Supranational Reference Laboratory and WHO Collaborating Centre in Tuberculosis Laboratory Strengthening (ITA-98). More details on access and application procedures can be found at https://apps.who.int/whocc/Detail.aspx?ySOkyLfD6Cp8kC9isuaZ/A==.
DNA quality and quantity assessment
The quality and quantity of extracted DNA were assessed using the Qubit Fluorometer (Thermo Fisher Scientific, USA) to ensure high-quality DNA suitable for subsequent analyses. The extracted genomic DNA was stored at −80 °C until further processing for library preparation and WGS.
Whole genome sequencing (WGS)
Whole genome sequencing (WGS) for isolates from Piedmont, Lombardy, and Emilia-Romagna and most of the Lazio strains was performed using the Nextera XT kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, genomic DNA was fragmented using enzymatic, followed by end-repair, adapter ligation, and PCR amplification. The resulting libraries were purified and quantified to ensure optimal library quality and concentration. Paired-end sequencing was conducted to generate short-read sequences with an average read length of 150 base pairs.
A portion of the isolates from Lazio were sequenced using IonTorrent sequencing platforms. DNA libraries were prepared according to the manufacturer’s instructions, including fragmentation, adapter ligation, and amplification, using the Ion Xpress Plus Fragment Library Kit (Thermo Fisher Scientific, USA) for sample library preparation. The Chef and S5 platforms were used for automated chip preparation and sequencing, respectively; the procedure resulted in the production of 250 bp reads.
Data analysis
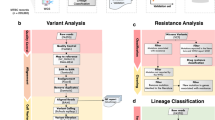
Sequence data were processed using the MTBseq pipeline for MTBC genomic analysis16. The pipeline includes modules for quality control, read mapping, variant calling, and annotation. Raw sequence reads were quality-filtered, trimmed, and aligned to M. tuberculosis H37Rv ATCC 27294 (NC_000962.3) as reference genome using established algorithms and software tools within the MTBseq framework. Variant calling was performed to identify single nucleotide polymorphisms (SNPs), insertions, deletions, and structural variations relative to the reference genome.
Drug resistance prediction
Drug resistance profiles were determined by analyzing mutations occurring in or upstream of genes linked to resistance against specific drugs as described previously17 and based on the 2023 WHO catalogue of mutations18. The WHO catalogue serves as a comprehensive reference for known genetic variants associated with drug resistance in MTBC strains, providing a standardized framework for drug resistance prediction. The use of this catalogue ensures that our approach is aligned with global standards and incorporates the most current and broadly recognized mutations associated with drug resistance. The drug resistance prediction algorithm implemented within the MTBseq pipeline (https://github.com/ngs-fzb/MTBseq_source)16 Importantly, we utilized the “--lowfreq_vars” option in MTBseq to enhance detection sensitivity for low-frequency variants. This setting allows MTBseq to consider minority alleles as potential resistant subpopulations, crucial for identifying mutations associated with drug resistance even at low frequencies.
Ethical considerations
This study involved the analysis of de-identified Mycobacterium tuberculosis complex isolates obtained from various health centers across Italy. Due to the retrospective nature of the study and the use of anonymized bacterial strains, which do not involve direct human subjects, a waiver of consent was granted. This waiver was approved by the Ethics Committee of the National Institute for Infectious Diseases “L. Spallanzani” - IRCCS, the coordinating center of the study. The Ethics Committee approval number is 46/2019.
Data Records
The full metadata, including run accessions, drug susceptibility profiles, HIV status, and additional relevant details, has been deposited in Zenodo under the https://doi.org/10.5281/zenodo.1478023813. This repository ensures long-term accessibility and provides a citable reference for future research. The raw sequence data from this study are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession numbers SRP53878319, SRP50914320, and SRP37328721. These data encompass high-quality whole genome sequences and provide a rich source for future analyses of MTBC genetic variability. Additionally, processed files, including variant calls, are accessible in Zenodo under the https://doi.org/10.5281/zenodo.1478023813,22,23.
Technical Validation
Quality control of DNA extraction
The quality and integrity of genomic DNA extracted from MTBC isolates were assessed using standardized protocols. We evaluated DNA purity and quantity using spectrophotometric measurements and gel electrophoresis. Samples with high-quality DNA (A260/A280 ratio ~1.8) and sufficient concentration (>10 ng/μL) were included in the study to ensure the reliability of downstream analyses.
Validation of library preparation
The efficiency and accuracy of DNA library preparation using the NExtera XT kit were validated through rigorous quality control measures. We assessed the size distribution and concentration of prepared libraries using automated electrophoresis (e.g., Agilent Bioanalyzer) and quantitative PCR (qPCR). Libraries with appropriate fragment size distribution and optimal concentration were selected for subsequent sequencing to ensure high-quality data generation. Validation of library preparation followed similar procedures when using the Ion Torrent system.
Sequencing quality control
Prior to data analysis, raw sequence reads obtained from Illumina and IonTorrent sequencing platforms underwent stringent quality control procedures. Low-quality reads were defined as those with a Phred quality score below 30 in more than 10% of bases or those with a read length shorter than 20 bp after trimming. Adapter sequences and PCR duplicates were removed using SAMtools v1.13. Base quality scores were assessed to ensure data integrity. Sequencing depth was considered adequate if the mean genome-wide coverage was ≥20×, ensuring sufficient depth for accurate variant calling and genomic analysis.
Validation of variant calling
The accuracy and reliability of variant calling were validated using reference materials and internal controls. Known reference strains with characterized genetic variants were included in sequencing runs to assess the sensitivity and specificity of variant detection. Furthermore, variant calls were compared against independent validation methods (e.g., Sanger sequencing) for selected genomic regions to confirm the accuracy of detected variants.
Reproducibility of data analysis
The reproducibility of data analysis using the MTBseq pipeline was evaluated through independent data processing and analysis runs. Multiple replicates of the same dataset were analysed using identical parameters and workflows to assess the consistency and reproducibility of results. Additionally, data analysis outcomes were validated by comparing results obtained from different computational tools and pipelines to ensure robustness and reliability. Since only one sample per patient was included in the dataset, technical or biological replicates were not part of the metadata table.
Validation of drug resistance prediction
Phenotypic drug susceptibility testing (pDST) for first-line antituberculosis drugs, as well as extended DST for fluoroquinolones and Group A drugs (bedaquiline and linezolid) in rifampicin-resistant or multidrug-resistant MTBC isolates, was performed as described previously16. The accuracy of drug resistance prediction was validated by comparing predicted drug resistance profiles with pDST results for a subset of MTBC isolates. Concordance between predicted and observed drug resistance phenotypes was assessed for first-line and second-line anti-tuberculosis drugs, providing validation of the predictive capability of the bioinformatics approach. Within this dataset, 94 (3.7%) isolates were identified as rifampicin-resistant (RR-TB), with 83 (88.3%) of these also showing resistance to isoniazid, thereby classifying them as multidrug-resistant TB (MDR-TB). Additionally, 22 (23.4%) of the MDR-TB strains exhibited resistance to fluoroquinolones, categorizing them as pre-extensively drug-resistant TB (pre-XDR-TB) (Table 1). In terms of resistance to newer and repurposed drugs, WGS analysis revealed three strains with mutations conferring resistance to linezolid and thirteen strains with mutations in the Rv0678 gene associated with resistance to bedaquiline and clofazimine; no mutations linked to resistance against delamanid were found.
Code availability
Different tools have been employed for data analysis, and the following sections describe their versions, settings, and parameters:
• FastQC (v0.11.3) with default parameters.
• MTBseq (v1.1.0) with the following parameters for drug resistance detection:
--mincovf 1 --mincovr 1 --minfreq. 5 --minphred20 1
and for comparative genomcis
--mincovf 4 --mincovr 4 --minfreq. 75 --minphred20 4
• RAxML-NG (v1.0.2) with with the following parameters: ‘–model GTR + G + ASC_LEWIS
• FigTree v1.4.4
• iTol online software.
References
Global tuberculosis report 2024. Geneva: World Health organization; licence: CC BY-NC-SA 3.0 IGO (2024).
Global tuberculosis report 2023. Geneva: World Health Organization; Licence: CC BY-NC-SA 3.0 IGO (2023).
Merker, M. et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet 47, 242–249, https://doi.org/10.1038/ng.3195 (2015).
Meehan, C. J. et al. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat Rev Microbiol 17, 533–545 (2019).
Yenew, B. et al. A smooth tubercle bacillus from Ethiopia phylogenetically close to the Mycobacterium tuberculosis complex. Nat Commun 14, 7519, https://doi.org/10.1038/s41467-023-42755-9 (2023).
Gagneux, S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol 16, 202–213, https://doi.org/10.1038/nrmicro.2018.8 (2018).
Allix-Béguec, C. et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med. 379(15), 1403–1415 (2018).
Lalor, M. K. et al. Utility of whole-genome sequencing in investigating a hospital outbreak of multidrug-resistant tuberculosis. Nature Communications 14, 2391 (2023).
Didelot, X. et al. Interpreting whole genome sequencing for investigating tuberculosis transmission: a systematic review. BMC Medicine 21, 179 (2023).
Sotgiu, G. et al. TB elimination: theory and practice in Europe. Eur Respir J. 43(5), 1410–1420 (2014).
Giannoni, F. et al. Decreasing trend of drug-resistant TB in Italy. Int. J. Tuberc. Lung Dis. 26, 775–783 (2022).
Villa, S. et al. Outbreak of pre- and extensively drug-resistant tuberculosis in Northern Italy: urgency of cross-border, multidimensional, surveillance systems. Eur Respir J 58, 2100839 (2021).
Ghodousi, A. Comprehensive Whole Genome Sequencing Dataset of Mycobacterium tuberculosis Strains Collected Across Italy [version v3]. Zenodo. https://doi.org/10.5281/zenodo.15125707 (2025).
Liu, Q. et al. Genetic diversity, evolution and transmission of Mycobacterium tuberculosis lineage 2. Front. Microbiol. 11, 1432 (2020).
Guerra-Assunção, J. A. et al. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife 4, e05166 (2015).
Kohl, T. A. et al. MTBseq: a comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates. PeerJ 6, e5895 (2018).
Ghodousi, A. et al. Evaluating the efficacy of whole genome sequencing in predicting susceptibility profiles for first-line antituberculosis drugs. Clinical Microbiology and Infection https://doi.org/10.1016/j.cmi.2024.09.024 (2024).
World Health Organization. Catalogue of mutations in Mycobacterium tuberculosis complex and their association with drug resistance, second edition. Geneva: World Health Organization; Licence: CC BY-NC-SA 3.0 IGO (2023).
Ghodousi, A. et al. Whole genome sequencing dataset of Mycobacterium tuberculosis strains from Italy. NCBI Sequence Read Archive. SRP538783 https://identifiers.org/ncbi/insdc.sra:SRP538783 (2024).
Ghodousi, A. et al. Evaluating the efficacy of whole genome sequencing in predicting susceptibility profiles for first-line antituberculosis drugs. NCBI Sequence Read Archive. SRP509143 https://identifiers.org/ncbi/insdc.sra:SRP509143 (2024).
National Institute of Infectious Diseases “L. Spallanzani”. Whole genome sequencing of Mycobacterium tuberculosis isolated from Tuberculosis cases in Italy. NCBI Sequence Read Archive. SRP373287 https://identifiers.org/ncbi/insdc.sra:SRP373287 (2022).
Ghodousi, A. Comprehensive Whole Genome Sequencing Dataset of Mycobacterium tuberculosis Strains Collected Across Italy [version v2]. Zenodo. https://doi.org/10.5281/zenodo.14808803 (2025).
Ghodousi, A. Comprehensive Whole Genome Sequencing Dataset of Mycobacterium tuberculosis Strains Collected Across Italy [version v1]. Zenodo. https://doi.org/10.5281/zenodo.14780239 (2025).
Acknowledgements
This work was supported by the Italian Ministry of health, through Ricerca Corrente Line n. 4, Project n. 1, and CCM-2018 project (Centro Nazionale per la Prevenzione ed il Controllo delle Malattie) “Definizione di strategie di controllo della tubercolosi associata ad HIV in Italia nel contesto di una strategia di eliminazione della malattia tubercolare” and for the analysis by CCM-2023 project (Centro Nazionale per la Prevenzione ed il Controllo delle Malattie) “Approccio integrato al rafforzamento del contrasto alla Tubercolosi: dalla diagnostica e gestione clinica. alla sanità pubblica”. Our sincere thanks go to Alma Zinola for her assistance with a part of Bioinformatics analysis.
Author information
Authors and Affiliations
Contributions
Arash Ghodousi (A.G.), Angela Cannas (A.C.), Elisa Tagliani (E.T.), Maryam Omrani (M.O.), Virginia Batignni (V.B.), Francesco Bisognin (F.B.), Emanuele Borroni (E.B.), Ornella Butera (O.B.), Luigi Ruffo Codecasa (L.R.C.), Paola Dal Monte (P.D.M.), Flavio De Maio (F.D.M.), Giovanni Delogu (G.D.), Giovanna Mattei (G.M.1), Giulio Matteo (G.M.2), Francesco Messina (F.M.), Cinzia Perilli (C.P.), Michela Sali (M.S.), Giovanni Sotgiu (G.S.), Marina Tadolini (M.T.), Stefania Torri (S.T.), Chiara Vismara (C.V.), Enrico Girardi (E.G.), Daniela Maria Cirillo (D.M.C.). Study design: D.M.C., A.G., E.T. and E.G. Development and methodology: D.M.C., A.G., E.T., E.B., A.C. and E.G. Data collection: A.G., A.C., E.T., M.O., V.B., F.B., E.B., O.B., L.R.C., P.D.M., F.D.M., G.D., G.M.1, G.M.2, F.M., C.P., M.S., G.S., M.T., S.T., C.V., E.G., D.M.C. Whole genome Sequencing: A.G., V.B., A.C., O.B., F.D.M. Quality control and data submission to public repository: A.G. and F.M. Data analysis and interpretation: A.G., M.O., E.T. and D.M.C. Technical Validation: A.G., O.M. and V.B. Writing all sections of the manuscript: A.G., A.C. and M.O. Draft revision: All authors. Manuscript final approval: D.M.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ghodousi, A., Cannas, A., Tagliani, E. et al. Comprehensive Whole Genome Sequencing Dataset of Mycobacterium tuberculosis Strains Collected Across Italy. Sci Data 12, 624 (2025). https://doi.org/10.1038/s41597-025-04966-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04966-1
