
Abstract
Fusarium neocosmosporiellum causes alfalfa root rot, leading to significant economic losses. To explore the genetics of F. neocosmosporiellum, strain CA18-1 was isolated from infected alfalfa roots and subjected to whole-genome analysis. A scaffold-level reference genome of strain CA18-1 was assembled using both Nanopore long-read and Illumina short-read sequencing technologies. The assembled genome measured 63,424,297 base pairs (bp) in size, with a contig N50 of 6,480,858 bp and a contig N90 of 3,230,245 bp. A total of 10,486,459 bp repeat sequences and 28,006 protein-coding genes were predicted, of which 15,389 were functionally annotated. This study enhances our understanding of the genetic basis for F. neocosmosporiellum’s pathogenicity, providing a foundation for future studies on disease control and fungal evolution.
Similar content being viewed by others

Background & Summary
Alfalfa (Medicago sativa L.) is a perennial, high-quality legume forage with diverse uses. It offers many advantages, including rich nutrient content, high biomass yield, strong stress resistance, and wide adaptability, making valuable both for feeding and ecological purposes1,2. As one of the most important forages in the world, alfalfa is widely planted in more than 80 countries, covering over 33 million square hectares3. In China, the rapid expansion of animal husbandry has led to a growing demand for high-quality alfalfa. However, intensive cultivation and continuous cropping have contributed to increased prevalence of diseases, pests, and weeds. These challenges significantly reduce the yield and quality of alfalfa, causing substantial losses to China’s animal husbandry. To date, 31 fungal diseases, four bacterial diseases, one viral disease, and one phytoplasma disease have been identified in alfalfa4. Among them, alfalfa root rot is an important root disease, causing annual yield losses of up to 40% and field mortality rate exceeding 60%5.
Alfalfa root rot is caused by a variety of pathogens, including bacteria, fungi, viruses and nematodes. The primary fungal pathogens responsible for this disease are Fusarium spp., Phytophthora spp., Rhizoctonia spp., and Aphanomyces spp. These pathogens can cause alfalfa root rot either individually or in combination5,6,7. Studies have shown that Fusarium is the predominant cause of root rot, with reports of up to 20 species or varieties of Fusarium pathogens associated with alfalfa root rot in the world5,8. Furthermore, new pathogen species continue to emerge. Environmental factors such as temperature9 and soil moisture10 can influence the diversity of pathogens, leading to variations under different ecological conditions. In China, Fusarium species causing alfalfa root rot are mainly F. oxysporum, F. solani, and F. acuminutum5. In Egypt, the primary pathogens are F. oxysporum, F. semitectum, and F. catenatum11, while in Canada, F. oxysporum, F. semitectum, and F. solani are the most prevalent12.
Fusarium is a fungal genus within phylum Ascomycota13. As one of the most significant plant pathogens, Fusarium can infect a variety of crops including maize, wheat, or corn, leading to major diseases14,15,16. Certain species produce mycotoxins during infection, causing food poisoning in humans and animals17. Additionally, some Fusarium species can directly infect humans and animals, resulting in serious diseases18,19. Fusarium is known for its high heterogeneity, making classification challenging. However, accurate species identification is crucial for biological, epidemiological, and toxicological research. Current identification methods rely on morphological characteristics and the DNA-based molecular approaches20,21.
In this study, F. neocosmosporiellum strain CA18-1 was isolated from infected alfalfa roots in China. This species is shown to be pathogenic to plants including soybean22, peanut23 and mango24. We sequenced the whole genome of strain CA18-1 using a combination of long-read sequencing and short-read sequencing. We evaluated the genome assembly, including coding genes, non-coding RNAs, and repetitive sequences, and annotated genes using various databases. This study provides insight into the genome structure and function of strain CA18-1, contributing to our understanding of its biological traits and pathogenic mechanisms.
Methods
Sample collection and extraction of genomic DNA
In this study, the alfalfa root rot fungal CA18-1 was isolated from diseased alfalfa root collected from fields in Arhorchin Banner in Chifeng, China (Fig. 1A,B). And the morphological characteristics align with those of F. neocosmosporiellum24 (Fig. 1C–E). The strain CA18-1 was inoculated in potato dextrose broth (PDB) and cultured at 180 rpm/min 25 °C for 5 days. The culture medium was filtered through four layers of sterile gauze to collect the mycelia. Genomic DNA was extracted from strain CA18-1 using a plant genome DNA extraction kit (Tiangen Biothech, Beijing, China). The DNA quality was assessed using Qubit (Thermo Fisher Scientific, Waltham, MA) and a Nanodrop (Thermo-Fisher Scientific, Waltham, MA).

Alfalfa (Medicago sativa L.) root rot disease and morphology of the fungal strain CA18-1. (A) Plant wilting. (B) Browning and decaying tissue at the plant stem base. (C) Top view of the colony cultured on potato dextrose agar (PDA) plates after seven days of incubation. (D) Microconidia. (E) Ascospores with ornate walls. Scale bars are 10 μm (D) and 20 μm (E).
Genome sequencing, data quality control and de novo assembly
Whole-genome sequencing of F. neocosmosporiellum strain CA18-1 was performed by Genedenovo Biotechnology Co., Ltd (Guangzhou, China) using both Oxford Nanopore PromethION (long-read) and Illumina NovaSeq X Plus (short-read) platforms. For Nanopore sequencing, a sequencing library was constructed using ONT’s library construction kit SQK-LSK110 (Oxford Nanopore Technologies, Oxford, UK). ABI StepOnePlus Real-Time PCR System (Life Technologies, CA, USA) was used to detect the quality of the library, and Agilent 2100 (Agilent, Santa Clara, CA) was used to evaluate the size of the inserted fragment, followed by ONT kit EXP-NBD104/114. Sequencing was performed on the Oxford Nanopore PromethION (Oxford Nanopore Technologies, Oxford, UK) platform. The average depth of sequencing achieved with Nanopore reads was 234X. As a result, a total of 1,449,861 reads were obtained with a mean read length of 13,076.1 bp and the reads N50 length of 22,698 bp (Table 1).
For Illumina sequencing, a sequencing library was constructed using the Illumina DNA Prep Kit (Illumina, CA, USA). A DNA library of 300-400 bp insert fragments was first prepared, and genome sequencing was subsequently performed using the Illumina NovaSeq X Plus platform (read length: 150 bp). The average depth for Illumina reads was 40X. The raw data were filtered using FASTP (version 0.20.0)25. After filtering, 18,235,894 reads were retained with 2,611,981,952 bp bases quality values reached Q20 and 2,548,654,550 bp bases quality values reached Q30 (Table 2).
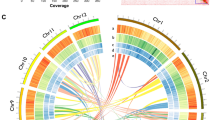
The clean reads from Nanopore sequencing were corrected using FMLRC (version 1.0.0)26. The corrected Nanopore sequencing reads were then reassembled using the Canu (version 2.2)27 software. To verify the assembly quality and determine the final genomic sequence, the Nanopore sequencing reads were aligned to the assembled genomic sequence using Racon (version 1.4.10)28, and the corrected genomic sequence was output. Additionally, the Illumina sequencing reads were aligned to the corrected Nanopore sequencing genomic sequence using Pilon (version 1.23)29, and the genomic sequence was corrected using the default software parameters. The corrected genomic sequence and the information about the correction sites were then output. The final assembly of the F. neocosmosporiellum strain CA18-1 genome is summarized in Table 3. A de novo scaffold-level genome measuring 63,424,297 bp was generated from 17 contigs, with a Contig N50 of 6,480,858 bp and a Contig N90 of 3,230,245 bp. The genome had a GC content of 49.76%, with the longest contig measuring 7,768,507 bp and the shortest contig measuring 41,112 bp (Fig. 2; Table 4).

Circular map of Fusarium neocosmosporiellum strain CA18-1 genome assembly. (a) Physical locations of 17 contigs. Bar = 1 kb. (b,c) Protein-coding genes on the forward and reverse strands, annotated with KOG classes. (d) Gene density. (e) Transposable element (TE) density. (f) Tanden repeat density.
Gene prediction and annotation
Coding genes represent the core functional regions of the genome of a species, encoding proteins necessary for the physiological and biochemical activities of the organism. Prior to gene prediction, the genome was subjected to repeat masking using BEDtools (version 2.28.0)30. The gene prediction analysis was then carried out on the repeat-masked genome utilizing Funannotate (version 1.8.9)31, resulting in a total of 28,006 predicted genes in the genome of F. neocosmosporiellum strain CA18-1 (Table 4). The total length of these genes was 51,444,891 bp, which accounted for 81.11% of the entire genome length. The distribution map of gene lengths showed that the largest number of genes fell within the >3000 bp range, totaling 2,159 genes (Fig. 3).

Length distribution of predicted protein-coding genes in Fusarium neocosmosporiellum strain CA18-1.
Non-coding RNAs, such as rRNAs, were predicted using RNAmmer (version 1.2)32, while tRNAs were identified using tRNAscan (version 1.3.1)33. Small RNAs (sRNA) and microRNAs (miRNA) were predicted through cmscan (version 1.1.2)34 by comparing with the Rfam database34. Analysis of the genomic data for F. neocosmosporiellum strain CA18-1 revealed the following: 345 tRNAs, 3 sRNAs, 0 miRNAs, 38 28s-rRNAs, 36 18s-rRNAs, and 65 5s-rRNAs (Table 5). Tandem repeats were predicted using Tandem Repeats Finder (TRF, version 4.09.1)35 software, while interspersed repeats were identified using EDTA (version 2.0.0)36. The results of the genome repeat sequence predictions for the sequenced strains are summarized in Table 6.
Our genome annotation pipeline employed an integrated strategy to maximize functional insights. Initial gene predictions underwent rigorous multi-database validation: assembled sequences were first queried against NCBI’s non-redundant database37 using BLASTp (e-value cutoff 1 × 10^-5) for broad functional classification, followed performed protein alignment with Swiss-Prot database38. And KOG39, KEGG40,41, and GO42 databases were used for metabolic pathway reconstruction. Subsequent domain characterization utilized Pfam-Scan (version 1.6) with the Pfam library43 (version 32.0), identifying conserved protein motifs unique to Fusarium species. Through this multi-tiered annotation pipeline, we functionally characterized 15,389 high-confidence unigenes (54.9% of the total 28,006 predicted genes), achieving comprehensive functional coverage with 98.36% of unigenes receiving at least one functional assignment through orthogonal evidence from multiple databases (Table 7).
Data Records
The Illumina and Nanopore sequencing data have been deposited in the NCBI Sequence Read Archive database under the SRA accession SRP56537044. The assembled genomes are available in GenBank under the accession number GCA_041296245.145. The genome annotation results have been deposited in the figshare46 database. The deposited CDS and protein fasta files contain only the 15,389 high-confidence unigenes with complete coding sequences. The gff file (CA18-1.gff) includes all 28,006 predicted gene models.
Technical Validation
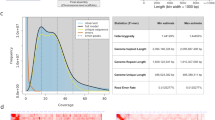
The Illumina short reads served as input for Jellyfish (version 2.3.017)47, from which a k-mer (k = 17) frequency distribution was obtained, as depicted in Fig. 4. After discarding the k-mers with abnormal depth, the genome size was estimated by using the formula genome size = k-mers number/average depth of k-mers. Consequently, the estimated genome size of the F. neocosmosporiellum strain CA18-1was 59.01 Mb, with a heterozygosity rate of 0.0807%. The assembly quality was evaluated based on contig length and BUSCO analysis. The N50 lengths of the primary assemblies exceeded 6.5 Mb, with the longest contig reaching 7.8 Mb. BUSCO (version 5.2.2)48 was used to assess the completeness of the genome assembly using the ascomycota dataset. Results of the genome quality assessment indicated that the assembly of CA18-1 was both complete and accurate, as single-copy orthologous genes in F. neocosmosporiellum strain CA18-1 matched 98.68% of all 758 complete core genes in the ascomycota dataset (Table 8).

k-mer (k = 17) frequency distribution generated using Illumina sequencing data of Fusarium neocosmosporiellum strain CA18-1. The x-axis shows k-mer depth (coverage), while the y-axis represents frequency. The primary peak at ~40X indicates the dominant homozygous genome component, with the minor peak at ~20X representing heterozygous regions (heterozygosity rate: 0.0807%).
Code availability
All parameters and options used in the study are described in the Methods. We used publicly available programs and not custom programs.
References
Bagavathiannan, M. V., Gulden, R. H., Begg, G. S. & Van Acker, R. C. The demography of feral alfalfa (Medicago sativa L.) populations occurring in roadside habitats in Southern Manitoba, Canada: implications for novel trait confinement. Environmental science and pollution research international 17, 1448–1459, https://doi.org/10.1007/s11356-010-0330-2 (2010).
Greene, S. L., Kesoju, S. R., Martin, R. C. & Kramer, M. Occurrence of transgenic feral alfalfa (Medicago sativa subsp. sativa L.) in alfalfa seed production areas in the United States. PloS one 10, e0143296, https://doi.org/10.1371/journal.pone.0143296 (2015).
Mielmann, A. The utilisation of lucerne (Medicago sativa): a review. British Food Journal 115, 590–600 (2013).
Chen, J., Guo, Z. W., Pan, C. Q., Xiang, P. & Liu, D. W. Research status of alfalfa diseases, insect pests and weeds. Journal of Grassland and Forage Science 01, 1–14 (2022).
Fang, X. L., Zhang, C. X. & Nan, Z. B. Research advances in Fusarium root rot of alfalfa (Medicago sativa). Acta Prataculturae Sinica 28, 169–183 (2019).
Mirhendi, H. et al. Preliminary identification and typing of pathogenic and toxigenic Fusarium species using restriction digestion of ITS1-5.8S rDNA-ITS2 region. Iranian journal of public health 39, 35–44 (2010).
Cong, L. L. et al. First report of root rot disease caused by Fusarium tricinctum on alfalfa in north China. Plant Disease 100, 1503 (2016).
Uddin, W. & Knous, T. R. Fusarium species associated with crown rot of alfalfa in Nevada. Plant Disease 75, 51–56 (1991).
Martin, F. N. & Loper, J. E. Soilborne plant diseases caused by Pythium spp.: Ecology, epidemiology, and prospects for biological control. Critical reviews in plant sciences 18, 111–181 (1999).
Kuan, T. & Erwin, D. Predisposition effect of water saturation of soil on Phytophthora root rot of alfalfa. Phytopathology 70, 981–986 (1980).
Elnasr, H. & Leath, K. T. Crown and root fungal diseases of alfalfa in Egypt. Plant Disease 67, 509–511 (1983).
Couture, L. et al. Fusarium root and crown rot in alfalfa subjected to autumn harvests. Canadian Journal of Plant Science 82, 621–624 (2002).
Lin, X. M., Li Z. Q. & Hou, J. Chinese fungi (China Agriculture Press, 2007).
Chang, H. et al. Isolation and Identification of Fusarium spp. from maize root rot in Gansu province. Journal of Maize Sciences 30, 184–190 (2020).
Ma, G. et al. Isolation, characterization, and pathogenicity of Fusarium species causing crown rot of wheat. Frontiers in microbiology 15, 1405115, https://doi.org/10.3389/fmicb.2024.1405115 (2024).
Lim, J. G. et al. Rapid and nondestructive discrimination of Fusarium asiaticum and Fusarium graminearum in hulled barley (Hordeum vulgare L.) using near-infrared spectroscopy. Journal of Biosystems Engineering 42, 301–313, https://doi.org/10.5307/JBE.2017.42.4.301 (2017).
de Oliveira Rocha, L. et al. Molecular characterization and fumonisin production by Fusarium verticillioides isolated from corn grains of different geographic origins in Brazil. International journal of food microbiology 145, 9–21, https://doi.org/10.1016/j.ijfoodmicro.2010.11.001 (2011).
Zhang, L. Y., Bao, M. L., Bao, H. L. & Wei, M. L. Production and prevention of mycotoxins in silage. Chinese Journal of Microecology 34, 1238–1241 (2022).
Chen, L., Qiu, M. & Dai, X. J. Research progress on the mechanism and animal models of keratitis caused by pathogen infection. Chinese Journal of Comparative Medicine 32, 137–144 (2022).
Singha, I. M., Kakoty, Y., Unni, B. G., Das, J. & Kalita, M. C. Identification and characterization of Fusarium sp. using ITS and RAPD causing fusarium wilt of tomato isolated from Assam, North East India. Journal, genetic engineering & biotechnology 14, 99–105, https://doi.org/10.1016/j.jgeb.2016.07.001 (2016).
Dumin, W., Han, Y. K., Park, J., Bae, Y. & Back, C. Identification and classification of pathogenic Fusarium isolates from cultivated Korean cucurbit plants. Korean Journal of Agricultural Science 49, 121–128 (2022).
Jimenez Madrid, A. M., Allen, T., & Wilkerson, T. First report of Neocosmospora stem rot of soybean Caused by Fusarium neocosmosporiellum in Mississippi. Plant disease, Advance online publication, https://doi.org/10.1094/PDIS-05-22-1200-PDN (2022).
Xu, M. et al. First report of peanut foot rot caused by Fusarium neocosmosporiellum in Shandong Province, China. Journal of Plant Pathology 103, 1059–1060 (2021).
Molina-Cárdenas, L. et al. Mango malformation disease caused by Fusarium neocosmosporiellum in Mexico. Canadian Journal of Plant Pathology 43, 714–721, https://doi.org/10.1080/07060661.2021.1880483 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics (Oxford, England) 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Wang, J. R., Holt, J., McMillan, L. & Jones, C. D. FMLRC: Hybrid long read error correction using an FM-index. BMC bioinformatics 19, 50, https://doi.org/10.1186/s12859-018-2051-3 (2018).
Koren, S. et al. M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research 27, 722–736, https://doi.org/10.1101/gr.215087.116 (2017).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome research 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England) 26, 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010).
Li, W. C. & Wang, T. F. PacBio Long-Read Sequencing, Assembly, and Funannotate Reannotation of the Complete Genome of Trichoderma reesei QM6a. Methods in molecular biology (Clifton, N.J.) 2234, 311–329, https://doi.org/10.1007/978-1-0716-1048-0_21 (2021).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic acids research 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Kalvari, I. et al. Non-Coding RNA Analysis Using the Rfam Database. Current protocols in bioinformatics 62, e51, https://doi.org/10.1002/cpbi.51 (2018).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome biology 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Li, W., Jaroszewski, L. & Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics (Oxford, England) 18, 77–82, https://doi.org/10.1093/bioinformatics/18.1.77 (2002).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 31, 365–370, https://doi.org/10.1093/nar/gkg095 (2003).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic acids research 28, 33–36, https://doi.org/10.1093/nar/28.1.33 (2000).
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for deciphering the genome. Nucleic acids research 32(Database issue), D277–D280, https://doi.org/10.1093/nar/gkh063 (2004).
Kanehisa, M. et al. From genomics to chemical genomics: new developments in KEGG. Nucleic acids research 34(Database issue), D354–D357, https://doi.org/10.1093/nar/gkj102 (2006).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29, https://doi.org/10.1038/75556 (2000).
Punta, M. et al. The Pfam protein families database. Nucleic acids research 40(Database issue), D290–D301, https://doi.org/10.1093/nar/gkr1065 (2012).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP565370 (2025).
Wang, L. Scaffold-level genome assembly of Fusarium neocosmosporiellum strain CA18-1. NCBI GenBank. https://identifiers.org/ncbi/insdc.gca:GCA_041296245.1 (2024).
Wang, L. The genome annotation of Fusarium neocosmosporiellum strain: CA18-1. figshare https://doi.org/10.6084/m9.figshare.28450001.v1 (2025).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics (Oxford, England) 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular biology and evolution 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Acknowledgements
This study was funded by the National Key Research and Development Program of China (2022YFD1300802).
Author information
Authors and Affiliations
Contributions
L.W.: Writing-Original Draf, Methodology, Formal Analysis. R.J.: Investigation, Formal Analysis. J.H.: Writing-Review & Editing, Methodology. Z.C.: Conceptualisation, Formal Analysis. X.Z.: Methodology. N.W.: Investigation. S.W.: Formal Analysis. Y.Z.: Conceptualisation, Writing-Review & Editing. K.L.: Funding Acquisition, Supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Jia, R., Hao, J. et al. Scaffold-level genome assembly of Fusarium neocosmosporiellum strain CA18-1. Sci Data 12, 762 (2025). https://doi.org/10.1038/s41597-025-05073-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05073-x
