
Abstract
Predatory insects serve as important biological control agents in ecosystems by efficiently reducing pest populations. Orius strigicollis is a highly effective beneficial predator in cultivated fields in China. It is widely used as a biological control agent against many pests, including thrips, aphids, and spider mites. The lack of public availability of O. strigicollis genomes has limited our ability to thoroughly explore and research its evolution and gene function, particularly genes related to pesticide resistance. Here, we report a chromosome-scale assembly of the O. strigicollis genome by PacBio High fidelity (HiFi) long-reads, high-throughput chromosome conformation capture (Hi-C) and Illumina sequencing technologies. The final genome assembly was 167 Mb with a scaffold N50 size of 12.49 Mb, and 97.87% of the assembly was anchored to 12 chromosomes. Annotation identified 11,551 protein-coding genes. In this article, we reported a high-quality chromosome-scale assembly genome for the species of the genus Orius for the first time, which provide valuable resources for functional gene research and genetic transformation of natural enemy insects.
Similar content being viewed by others

Background & Summary
Orius species are common natural enemies in farmland ecosystems and are relatively easy to mass produce. They have been developed as commercially available biological control agents and are widely used in greenhouse crops1,2,3. Orius species are commonly found scurrying about on the leaves and blossoms of various plants, including chili peppers, eggplants, flowering plants, and so on. Both the nymphs and adults of Orius display a polyphagous predatory behavior, and are proficient in preying upon a wide range of arthropod pests, including thrips, aphids, whiteflies, mites, moth eggs, and young caterpillar larvae4,5,6. In the absence of prey, they also feed on plant sap and pollen. Currently, Orius species are widely used to control thrips in vegetable cash crops, often in combination with other methods for comprehensive pest management7. In recent years, the widespread use of broad-spectrum insecticides has accelerated resistance development in pest populations. Moreover, the misuse and overreliance had a negative impact on the viability and reproduction of Orius, reducing their effectiveness in pest control8,9. Unfortunately, the dearth of comprehensive genomic resources has hindered in-depth research on Orius. As of now, merely two genomes of the Orius genus can be found in GenBank, and the assembly quality of these genomes leaves much to be desired. This shortcoming has placed significant restrictions on research into the insecticide-resistance mechanisms and potential resistance targets of Orius insects.
The predatory flower bug, Orius strigicollis (Poppius) (Hemiptera, Anthocoridae), is one of the dominant Orius species across southern provinces in China, such as Zhejiang, Guizhou, and Hainan10,11. At present, O. strigicollis has been developed as a biological control product and has shown encouraging results in controlling various thrips species6,8. This biological control method is part of an integrated pest management (IPM) strategy designed to reduce reliance on chemical pesticides. However, the effectiveness of O. strigicollis as a biological control agent is often insufficient in large-scale agricultural pest infestations (outbreaks), making the use of chemical agents necessary for effective pest control. However, the use of chemical pesticides significantly increases the mortality rate of O. strigicollis and drastically reduces its reproductive capacity8,12, undermining the stability and sustainability of these pest management strategies.
In order to promote the molecular biology research of natural enemy insects of the Orius genus and fill the gap in the realm of insecticide resistance of these natural enemy insects, this study endeavors to reveal the genome map of O. strigicollis through high-quality genome sequencing, assembly, and annotation analysis. This will provide a foundation for future research on the molecular mechanisms of insecticide resistance, help identify key resistance genes in O. strigicollis, and offer essential genetic resources for gene editing to enhance resistance in natural enemy insects.
Methods
Insects rearing and sample collection
Orius strigicollis populations were collected from eggplant (Solanum melongena L.) in Jiaxing (120.42° E, 30.45° N), Zhejiang, China, in 2023. These populations were maintained in an artificial climate chamber set at 26 ± 1 °C with a photoperiod of 14 hours light and 10 hours dark (L/D = 14:10). For the reproduction of O. strigicollis, western flower thrips (Frankliniella occidentalis) were provided as prey, and fresh bean pods (Phaseolus vulgaris) served as the oviposition and feeding substrate.
Genomic DNA
For Illumina sequencing, approximately 200 fifth instar nymphs of O. strigicollis were collected to extract genomic DNA (gDNA). The nucleic acid purity (OD260/280 and OD260/230), concentration, and nucleic acid absorption peak of nucleic acid were detected by Nanodrop 2000C (Thermo Fisher Scientific, USA). Qubit 3.0 was used to accurately detect genomic DNA concentration and compared with Nanodrop results to assess sample purity. Further, the genomic DNA integrity were detected by 1.5% agarose gel electrophoresis. After the nucleic acid sample was qualified, the library was constructed for Illumina sequencing. Subsequently, the gDNA underwent fragmentation to achieve 300~400 bp size, and a paired-end library was constructed following the Illumina standard protocol. Sequencing was conducted by Wuhan OneMore Tech Co., Ltd. on Illumina NovaSeq6000 sequencing platform, and a total of 30.06 Gb raw data was obtained (Table 1).
For long-read sequencing, approximately 200 fifth instar nymphs of O. strigicollis were collected and used to extract the gDNA. The purity, quantity, and integrity of the gDNA was assessed using Nanodrop 2000C (Thermo Fisher Scientific, USA), Qubit 3.0, and agarose gel electrophoresis. After the nucleic acid sample was qualified, a SMRTbell library was established to obtain the PacBio High fidelity (HiFi) long-reads using the SMRTBell template preparation kit 1.0 (PacBio) according to the product instructions with a fragment size of 20 kb. Next, Circular Consensus Sequence (CCS) mode was used to perform sequencing in PacBio Sequel II system. Finally, a sum of 11.61 Gb raw data was generated (Table 1).
For Hi-C sequencing, the gDNA was extracted from about 200 fifth instar nymphs of O. strigicollis and utilized to construct a Hi-C library following the standard procedure. Briefly, paraformaldehyde treated DNA was digested with DpnII to create sticky ends, then repaired and biotin-labeled. DNA fragments were ligated, cross-links removed with protease, purified, and fragmented into 300~500 bp. After that, ligation junctions were isolated through binding to streptavidin beads and prepared for sequencing by Wuhan OneMore Tech Co., Ltd. on Illumina NovaSeq6000 sequencing platform. A total of 91.17 Gb raw Hi-C data were obtained (Table 1).
For transcriptome sequencing, total RNA was extracted from 20 adults of O. strigicollis with nine repetitions using TRIzol reagent (Invitrogen), and then Nanodrop 2000C, Qubit 3.0, and agarose gel electrophoresis were use to evaluate the purity, quantity, and integrity of RNA. The cDNA library was constructed and sequenced using an Illumina Novaseq 6000 sequencing platform at Wuhan OneMore Tech Co., Ltd. Afer that, Qubit 2.0 and Agilent 2100 were used to assessed the library’s concentration and insert size, while Q-PCR (effective library concentration > 2 nM) precisely measured its effective concentration to ensure quality. A total of 32.43 Gb RNA-seq raw data was obtained for predicting protein-coding genes within the O. strigicollis genome (Table 1).
Genome assembly
The raw data of Illumina sequencing were strictly controlled and filtered by fastp (v 0.23.2) software13. The specific quality filtering criteria were as follows: (1) sequences contaminated with linkers longer than 5 bases were removed (>5 bp of adapter sequence); (2) sequencing fragments of low quality, defined as having more than 15% of bases with a mass value below 19 (≥15% of bases with Q-values < 19), were excluded; (3) reads with unknown base content exceeding 5% were discarded (N bases > 5%); (4) and sequences from paired ends were eliminated if affected by the aforementioned criteria. Subsequently, GCE (v 1.0.0)14 were employed for genome analysis and K-mer analysis. The genome size of O. strigicollis has been estimated at 187 Mb, with 29.95% repetitive sequences and exhibiting a heterozygosity rate of 1.38% at K = 17 (Table S1 and Fig. S1).
The Hifiasm v0.19.6 (https://github.com/chhylp123/hifiasm) and the purge_haplotigs algorithms (v1.0.4)15 were employed to assembly the initial contigs in HiFi reads with the default settings. In addition, Hi-C analysis was performed to further scaffold the genome assembly into chromosome-scale. We employed the HiCUP pipeline (v0.7.2)16 to generate a reliable and non-redundant contig interaction matrix, subsequently anchoring the contigs onto chromosomes using the 3D-DNA pipeline (https://github.com/theaidenlab/3d-dna). The Juicebox Assembly Tools17 were employed for manual error correction to address instances of chromosome inversion and translocation.
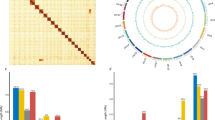
In total, the original 73 contigs interrupted the wrong contigs according to the interaction heatmap (Fig. 1), and sorted them. Finally, 12 chromosomes and 44 scaffolding were constructed (Fig. 2), with a maximum contig N50 length of 12.05 Mb and a maximum scaffold N50 length of 12.49 Mb (Table 2), in which the chromosome anchoring rate was 97.87% (assembly size = 167 Mb). In addition, BUSCO alignment analysis of assembly based on the hemiptera_odb10 database revealed that 2,468 (98.33%) BUSCO genes were complete (Table 2). After that, the chromosome and genome interaction maps of the species were constructed respectively, which results suggest the anchored genome assembly is complete and high-quality, suitable for detailed analyses.

Hi-C interactive heatmap of 12 chromosomes in Orius strigicollis genome.

Contig distribution maps for chromosomes of Orius strigicollis. The bars in grey represent entire lengths of chromosomes, and the contig numbers and the sizes of chromosomes were shown behind the bars.
Genome annotation
Repeat elements in the O. strigicollis genome were identified using homologous prediction and ab initio prediction methods in TRF (v4.09)18, RepeatMasker (open-4.0.9) (https://www.repeatmasker.org/), and RepeatModeler (open-1.0.11)19 software. There were 30.98% repeat elements of the genome, including DNA elements (11.6%), LINEs (2.3%), SINEs (0.46%), LTRs (1.69%), Satellite (1.4%), Simple_repeat (0.54%), and Unclassified (14.97%) (Table 5). Genome circos plot was generated by Circos (0.69–9). The distribution of genomic elements in O. strigicollis was revealed in the circos plot (Fig. 3). Utilizing the repeat-masked genome, the prediction of protein-coding genes was approached through homologous prediction (miniprot 0.11-r23420 & Liftoff 1.6.321), ab initio prediction (AUGUSTUS 3.3.222 & Genscan23) and RNA-sequencing (HISAT2 2.1.024, Stringtie 1.3.525 & TransDecoder (https://github.com/TransDecoder/TransDecoder)) with default settings. Then, the homologous gene prediction of O. strigicollis based on the genome and version accession of Cimex lectularius (GenBank assembly accession: GCF_000648675.2_Clec_2.1), Apolygus lucorum (GenBank assembly accession: GCA_009739505.2_ASM973950v2), Rhodnius prolixus (GenBank assembly accession: GCA_000181055.3_RproC3) and Nezara viridula (GenBank assembly accession: GCA_928085145.1_PGI_NEZAVIv3). Based on that, four gene model predictions were unite by MAKER2 (v2.31.10)26. Finally, total 11,551 protein-coding genes were identified according to HiFAP (Wuhan OneMore Tech Co., Ltd., https://www.onemore-tech.com/) with merging and eliminating redundancy (Table 3).

Genome characteristics of Orius strigicollis (window size 50 kb). The tracks indicate (a) GC content, (b) gene density, (c) repeat density, (d) LTR retroelement density, (e) LINE density, and (f) DNA transposons density.
The protein-coding genes were annotated based on NR (NCBI nonredundant protein), SwissProt (http://www.gpmaw.com/html/swiss-prot.html), TrEMBL (http://www.uniprot.org), KOG (eukaryotic orthologous groups of proteins), TF (Transcription Factor), InterPro, GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), and Pfam database. A total of 10,484 genes (91.61%) were well annotated with at least one database (Fig. 4). In addition, Rfam (v14.8)27 and miRBase28 databases were used to predict miRNAs, rRNAs and snRNAs. tRNAscan-SE (v1.3.1)29 was employed to predict tRNAs. In total, there were 38 miRNAs, 1011 tRNAs, 1334 rRNAs, and 81 snRNAs identified in O. strigicollis genome (Table 4).

The number of genes annotated in databases.
Data Records
The raw sequencing dataset of O. strigicollis in this study can be achieved from Sequence Read Archive (SRA) under PRJNA117627330 and SRP54026431, including WGS Illumina sequencing data, Pacbio HiFi sequencing data, Hi-C sequencing data and RNA sequencing data. The assembled genome of O. strigicollis was deposited at GenBank under accession JBJYUV00000000032. Additionally, the genome assembly and annotated genes have been made available in the Figshare repository33.
Technical Validation
The genome size of O. strigicollis was estimated to be 187 Mb, while the final genome assembly measured 167 Mb, and the scaffold N50 reached 12.49 Mb. Comparing the long-read data with the assembled genome reveals a comparison rate of 99.48% and a coverage rate of 99.95%. The short reading data were compared with the assembled genome, and the comparison rate was 93.51%, and the coverage rate was 99.95% (Table S2). The Hi-C heatmap displayed an organized pattern of chromosomal interactions, indirectly confirming the accuracy of the chromosome assembly (Fig. 1). The complete alignment of BUSCO genes accounted for 98.33% and was successfully identified in the O. strigicollis genome assembly (Table 2). In contrast to the scaffold-level assembly of O. laevigatus (2,050 scaffolds) and the contig-level assembly of O. insidiosus (4,518 contigs) (Table 6), our chromosome-level O. strigicollis genome, featuring 12 anchored chromosomes, may serve as the foundational reference genome for future Orius genus research.
Code availability
Data analysis was conducted with default parameters in this study unless otherwise specified in the Methods section, and no custom code was developed specifically for these analyses.
References
Atanasova, B. et al. Use of Orius laevigatus to control Frankliniella occidentalis (thysanoptera: thripidae) population in greenhouse pepper. Journal of Agriculture and Plant Sciences 20, 15–21 (2022).
Wang, J. et al. Control effects of Orius sauteri on Frankliniella occidentalis in pepper and eggplant flowers in greenhouses. Chinese Journal of Biological Control 39, 264 (2023).
Dai, X. et al. Control effect and field application of four predatory Orius species on Megalurothrips usitatus (Thysanoptera: Thripidae). J Econ Entomol. 117, 448–456 (2024).
Ali, S. et al. Using a two-sex life table tool to calculate the fitness of Orius strigicollis as a predator of Pectinophora gossypiella. Insects. 11, 275 (2020).
Ur Rehman, S. et al. Predatory functional response and fitness parameters of Orius strigicollis Poppius when fed Bemisia tabaci and Trialeurodes vaporariorum as determined by age-stage, two-sex life table. PeerJ 8, e9540 (2020).
Ding, H. et al. Integrating demography, predation rate, and computer simulation for evaluation of Orius strigicollis as biological control agent against Frankliniella intonsa. Entomol Gen 41, 179–196 (2021).
Lin, Q. et al. Improved control of Frankliniella occidentalis on greenhouse pepper through the integration of Orius sauteri and neonicotinoid insecticides. J Pest Sci 94, 101–109 (2021).
Lin, T. et al. Compatibility of six reduced-risk insecticides with Orius strigicollis (Heteroptera: Anthocoridae) predators for controlling Thrips hawaiiensis (Thysanoptera: Thripidae) pests. Ecotoxicol Environ Saf 226, 112812 (2021).
Rahman, M. et al. Residual selectivity of some pesticides on the predatory bug, Orius minutus (Hemiptera: Anthochoridae). Biocontrol Sci Technol 32, 467–483 (2022).
Yu, C. et al. The predatory bug Orius strigicollis shows a preference for egg-laying sites based on plant topography. PeerJ 9, e11818 (2021).
Zhang, C. et al. Establishment of a faba bean banker plant system with predator Orius strigicollis for the control of thrips Dendrothrips minowai on tea plants under laboratory conditions. Insects 12, 397 (2021).
Lin, T. et al. Toxicity risk assessment of flupyradifurone for the predatory pirate bug, Orius strigicollis (Poppius) (Heteroptera: Anthocoridae), a biological control agent of Diaphorina citri Kuwayama (Hemiptera: Liviidae). Ecotoxicol Environ Saf 267, 115632 (2023).
Chen, S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Liu, B. et al. Estimation of genomic characteristics by analyzing K-mer frequency in de novo genome projects. Quant. Biol. 35, 62–67 (2013).
Roach, M. et al. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Research (2015).
Neva, C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems (2016).
Benson, G. et al. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Price, A. et al. De novo identification of repeat families in large genomes. Bioinformatics 21, 351–358 (2005).
Shumate, A. et al. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021).
Slater, C. et al. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 1–11 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, 435–439 (2006).
Burge, C. & Samuel, K. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268, 78–94 (1997).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Holt, C. & Mark, Y. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 1–14 (2011).
Griffiths-Jones, S. et al. Rfam: annotating non-codingRNAs in complete genomes. Nucleic acids research 33, 121–124 (2005).
Kozomara, A., Birgaoanu, M. & Griffiths-Jones, S. miRBase: from microRNA sequences to function. Nucleic Acids Res 47, 155–162 (2019).
Lowe, T. & Sean, R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
NCBI Bioproject https://identifiers.org/ncbi/bioproject:PRJNA1176273 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP540264 (2025).
Wang, Y. Chromosome-level genome assembly of the predatory flower bug Orius strigicollis. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_046618645.1 (2025).
The genome annotation files for the predatory flower bug Orius strigicollis. Figshare. Collection. https://doi.org/10.6084/m9.figshare.28039496.v1 (2025).
Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFD1401200), the Major Science and Technology Projects in Xinjiang (2023A02009), Zhejiang High-level Talents Special Support Program (2023R5249), the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2022C04029; 2022C04016).
Author information
Authors and Affiliations
Contributions
Y.R.W. and Y.B.L designed the project. J.H., Z.J.Z., J.M.Z.,W.Y.D. and S.X.Z coordinated the study. L.M.C., L.Q.W. and S.Y.Z. conducted the sampling and sequencing; Y.R.W. analyzed the data; Y.B.L and X.W.L. got the funding; Y.R.W., F.U. and X.W.L. wrote the original draf and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Chen, L., Ullah, F. et al. Chromosome-level genome assembly of the predatory flower bug Orius strigicollis. Sci Data 12, 820 (2025). https://doi.org/10.1038/s41597-025-05149-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05149-8
