
Abstract
The marbled orb-weaver spider, Araneus marmoreus (Araneae: Araneidae), is distinguished by its unique inflated, pumpkin-like abdomen. Numerous genome studies have been conducted on Araneidae species, providing insights into their unique biological traits. However, studies on A. marmoreus remain limited, despite its ecological significance and intriguing morphology. The lack of a high-quality reference genome has further hindered in-depth exploration of its evolutionary biology and ecological dynamics. Here, we present a chromosome-level genome assembly for A. marmoreus, generated using a combination of Illumina, PacBio, and Hi-C sequencing technologies. The assembled genome is 2.39 Gb in size, comprising 13 chromosomes, with a scaffold N50 of 181.8 Mb and a contig N50 of 721.3 kb. The assembly achieved a BUSCO completeness score of 97.1% (n = 2,934), including 91.0% complete and single-copy BUSCOs and 6.1% complete and duplicated BUSCOs. Repetitive sequences accounted for 59.25% of the genome, and 23,381 protein-coding genes were annotated. This high-quality genome provides a valuable resource for advancing research into the evolutionary genomics and ecological dynamics of A. marmoreus.
Similar content being viewed by others

Background & Summary
Spiders, as predatory arthropods, exhibit an extraordinary diversity, with more than 136 families and 52 thousand extant species described to date1. One of the largest spider family, Araneidae is particularly notable, known as orb-web weaving spiders, comprising more than 3.1 thousand species globally1 and considered one of the species-rich groups of spiders2. Most members of Araneidae heavily rely on their orb-web, a multifunctional tool for prey capture, communication, courtship, and mating3,4,5 and have been central to research on spider silk6,7, web-building behaviors4 and sexual size dimorphism8.
The marbled orb-weaver, Araneus marmoreus is characterized by the female’s inflated, pumpkin-like abdomen (Fig. 1). Adults display vibrant yellow to orange coloration, with black markings and banded legs. The species spins typical large orb-webs (>50 cm) between tall grasses, shrubs, and forest edges. Their life cycle is seasonal: adults mate in late summer, lay eggs, and perish after the breeding season9,10.

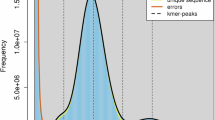
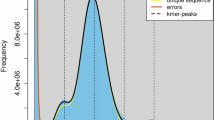
The genome analysis of orb-weaver spider Araneus marmoreus. (A) The survey analysis of A. marmoreus. (B) Heatmap of chromosome interactions in A. marmoreus. (C) Circos plot of distribution of the genomic elements in A. marmoreus. The inner ring contains a picture of orb-weaver spider. The outer rings of the circle represent means bellow, respectively: Chr: chromosomes, Gene: distribution of genes, GC: GC content; SINE: short interspersed nuclear element, LINE: long interspersed nuclear elements, LTR: long terminal repeat, DNA: DNA transposable elements. (D) Genomic synteny between A. marmoreus and Trichonephila antipodiana.
Up to now, although the genomes of 15 Araneidae spiders have been sequenced, this is insufficient for in-depth research on Araneidae spiders. The genus Araneus, in particular, lacks a chromosome-level genome. To fill this gap, we assembled a chromosome-level genome of A. marmoreus using PacBio HiFi, Illumina, and Hi-C sequencing technologies. The genome was annotated to identify repetitive elements, non-coding RNAs, and protein-coding genes. This high-quality genome provides a valuable foundation for further studies on Araneidae evolution and the genetic basis of orb-weaving spiders’ adaptations.
Methods
Sample collection and sequencing
The female specimens of Araneus marmoreus were collected from Chifeng City, Inner Mongolia Autonomous Region, China. The cephalothorax of the spiders, excluding the abdomen, was used for Illumina and PacBio sequencing, while leg muscle tissue was utilized for Illumina RNA-seq and Hi-C sequencing. The samples were first ground in liquid nitrogen, stored on dry ice, and subsequently sent to Berry Genomics (Beijing, China) for sequencing. Genomic DNA was extracted using the Qiagen Blood & Cell Culture DNA Mini Kit following the manufacturer’s protocol, optimized for PacBio and Illumina sequencing. PacBio sequencing employed Sequel II libraries with a 15 kb insert size, prepared using the SMRTbell™ Template Prep Kit 1.0-SPv3. Paired-end reads (150 bp) were generated using the Illumina NovaSeq platform for genome survey analysis and Hi-C sequencing. Total RNA was extracted from an adult female A. marmoreus using TRIzol (Invitrogen, USA) according to the manufacturer’s instructions and sequenced on the Illumina NovaSeq platform. We totally obtained 609.33 Gb clean data, including 149.68 Gb illumina reads (65×), 246.85 Gb Pacbio reads (107×), 204.81 Gb Hi-C reads (86×), and 7.99 Gb RNA reads (Table 1).
Survey analysis
First we used the “clumpify.sh” and “bbduk.sh” tools of BBTools suite v38.6711 to filter the Illumina reads. Then the filtered reads were feed to the “khist.sh” tool to estimate the k-mer distribution. And the software of GenomeScope v1.0.012 was used to calculate genome size with the maximum k-mer coverage cutoff was set to 10,000 and the k-mer sets 19. For results, the estimated genome size of A. marmoreus was 2.24 Gb, the heterozygosity was 0.94% (Fig. 1A).
Genome assembly
We used the softwere of Flye v2.513 to assembly the draft assemble genome through the PacBio long reads with minimum overlap between reads (-m) set to 3000. Then the software of Purge Haplotigs v1.1.014 was used to remove the heterozygous regions from the draft assembly genome. Next, the softwere of NextPolish v1.0.515 was used to polished the assembly genome from last step with Illumina reads. And the software of Minimap2 v2.1216 was used to align the reads with the assembly. Finally, the software of Juicer v1.6.217, 3D-DNA v.18092218, and Juicebox were used to obtain the chromosome-level assembly with Hi-C reads. In additation, we also remove the potential contaminants in the chromosome-level assembly through blast the NCBI nucleotide and UniVec databases with the software of HS-BLASTN19 and BLAST + (blastn) v2.7.120. And the software of BUSCO v5.2.221 pipeline was used to value the genome completeness with the arachnida_odb10 database (n = 2,934). In total, we obtained the chromosome assembly genome of A. marmoreus with the genome size of 2.39 Gb, scaffold N50 was 181.79 Mb, and contig N50 was 721.29 kb. A total of 13 chromosomes were assembled (Fig. 1B, Table 2), each larger than 100 Mb, and the Hi-C sequence was attached to the chromosome at a rate of 99.7%. The assembly achieved a BUSCO completeness score of 97.1% (n = 2,934), including 91.0% complete and single-copy BUSCOs and 6.1% complete and duplicated BUSCOs.
RNA assembly
The clean RNA illumina reads were mapped to the assembly genome by the HISAT2 v 2.2.022. Then using the Stringtie v2.1.323 to assemble the transcripts.
Genome annotation
Before genome annotation, the repetitive elements of the genome was first identified and softmasked by the software of RepeatModeler v2.0.124 and RepeatMasker v.4.1.425 through ab initio and homology-based searching with the Dfam database and RepBase RepeatMasker Edition database. In total, about 59.25% of assembly genome was annotated as repetitive elements, including 10.93% of DNA transposon elements, 3.21% of long terminal repeats (LTRs), 0.18% of long interspersed nuclear elements (LINEs), 0.18% short interspersed nuclear elements (SINEs), 41.48% of unclassified elements, 0.03% small RNAs, 0.01% satellites, 0.61% simple repeats, and 0.15% low-complexity regions (Table 3, Fig. 1C).
For gene structure annotation, we used the maker v3.01.0426 pipline based on ab-initio, EST and homologous proteins evidence. For ab-initio prediction, the software GeneMark-ETP v4.68_lic93 and Augustus v3.5.027 were employed for initially trained using the BRAKER v3.0.228. For EST evidence, the RNA transcripts were fed to maker pipline via the “est” option. For protein homology-based evidence, we downloaded the protein sequences of Bombyx mori (GCA_030269925.2), Drosophila melanogaster (GCA_000001215.4), Parasteatoda tepidariorum (GCA_000365465.3), Stegodyphus mimosarum (GCA_000611955.2) from NCBI, and Trichonephila antipodiana from GigaDB. And the proteins was fed to the maker pipline via the “protein” option. As the results, 23,381 protein-coding genes were identified, with an average length of 28,771.1 bp. Each gene exhibited an average of 6.91 exons, 6.75 CDS. The proteins annotated achieved a BUSCO completeness score of 97.8% (n = 2,934), including 85.8% complete and single-copy BUSCOs and 12% complete and duplicated BUSCOs.
For gene function annotation, the software EggNOG-mapper v2.1.1029, Diamond v2.0.14.15230, and InterProScan v5.48–83.031 were used to identify gene ontology (GO), expression coherence (EC), Kyoto Encyclopedia of Genes and Genomes pathways (KEGG), KEGG orthologous groups (KOs), and clusters of orthologous groups (COG) through eggNOG v5.032 based on the CDD33, Gene3D34, Panther35, Pfam36, and Superfamily37 databases. In total, 22,737 (97.25%) genes were identified with functional annotations. As a result, 16,690 genes were annotated with GO terms, and 13,891 genes were annotated at least one KEGG pathway (Table 4).
The software of Infernal v1.1.438 and tRNAscan-SE v2.0.939 were used to identified the Non-coding RNAs (ncRNAs) and transfer RNAs (tRNAs). The analysis revealed a total of 9,818 ncRNAs in the A. marmoroide genome, including 12,193 tRNAs, 2,997 ribosomal RNAs, 60 snoRNA, 47 microRNAs, 544 small nuclear RNAs, 35 ribozymes, and 330 other ncRNAs (Table S1).
Data Records
The raw data used in the manuscript including Illumina, PacBio, Hi-C, transcriptome and the genome assembly and annotation of Araneus marmoreus have been deposited at the ScienceDB (https://cstr.cn/31253.11.sciencedb.19518)40, and NCBI database with project number of PRJNA774480, BioSample number of SAMN23402377, genome number of GCA_050042785.1 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_050042785.1/)41, and SRA number of SRR32918500, SRR32918501, SRR32918502, and SRR32918503 (https://identifiers.org/ncbi/insdc.sra:SRP575255)42.
Technical Validation
The mapping reads for DNA and RNA illumina reads to the assembly genome were 93.90% and 82.40%. And the mapping rates of Hi-C sequence to the chromosome was 99.7%. The assembly completeness of BUSCO was 97.1% (n = 2,934), and the annotated proteins completeness of BUSCO was 97.8% (n = 2,934). We checked the synteny block between Araneus marmoreus and Trichonephila antipodiana of Araneidae (Fig. 1D), which showed that the A. marmoreus genome has a good genome synteny relationship with T. antipodiana. And we did the consensus quality (QV) values analysis by the software Merqury43 for evaluation of the assembly genome quality based on the illumina data, and the value was 36.8084.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
World Spider Catalog. World Spider Catalog. Version 25.5. Natural History Museum Bern, online at http://wsc.nmbe.ch, accessed on {3 Jan. 2025} (2025).
Kulkarni, S., Wood, H. M. & Hormiga, G. Advances in the reconstruction of the spider tree of life: A roadmap for spider systematics and comparative studies. Cladistics, https://doi.org/10.1111/cla.12557 (2023).
Hu, X. et al. Molecular mechanisms of spider silk. Cell Mol Life Sci 63, 1986–1999, https://doi.org/10.1007/s00018-006-6090-y (2006).
Vollrath, F. & Selden, P. The Role of Behavior in the Evolution of Spiders, Silks, and Webs. Annual Review of Ecology. Evolution, and Systematics 38, 819–846, https://doi.org/10.1146/annurev.ecolsys.37.091305.110221 (2007).
Fischer, A., Schulz, S., Ayasse, M. & Uhl, G. Pheromone communication among sexes of the garden cross spider. Sci Nat-Heidelberg 108, https://doi.org/10.1007/s00114-021-01747-9 (2021).
Arakawa, K. et al. 1000 spider silkomes: Linking sequences to silk physical properties. Science advances 8, eabo6043, https://doi.org/10.1126/sciadv.abo6043 (2022).
Correa-Garhwal, S. M., Babb, P. L., Voight, B. F. & Hayashi, C. Y. Golden orb-weaving spider (Trichonephila clavipes) silk genes with sex-biased expression and atypical architectures. G3 11, https://doi.org/10.1093/g3journal/jkaa039 (2021).
Kuntner, M. & Coddington, J. A. Sexual Size Dimorphism: Evolution and Perils of Extreme Phenotypes in Spiders. Annu Rev Entomol 65, 57–80, https://doi.org/10.1146/annurev-ento-011019-025032 (2020).
Fasola, M. & Mogavero, F. Structure and habitat use in a web - building spider community in northern Italy. Bolletino di zoologia 62/2, 159–166, https://doi.org/10.1080/11250009509356064 (1995).
Herbert W. L., Lorna R. L., Nicholas S. Spiders and Their Kin. St. Martin’s Press (2001).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nature Methods 17, 1103–1110 (2020).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Chen, Y., Ye, W., Zhang, Y. & Xu, Y. High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Research 43, 7762–7768 (2015).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Molecular Biology and Evolution 35, 543–548 (2017).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295 (2015).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna. 6, 11 (2015).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research 32, W309–W312 (2004).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics and Bioinformatics 3, lqaa108 (2021).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular Biology and Evolution 38, 5825–5829 (2021).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Finn, R. D. et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Research 45, D190–D199 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47, D309–D314 (2019).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45, D200–D203, https://doi.org/10.1093/nar/gkw1129 (2017).
Lewis, T. E. et al. Gene3D: Extensive prediction of globular domains in proteins. Nucleic Acids Res 46, D435–D439, https://doi.org/10.1093/nar/gkx1069 (2018).
Mi, H., Poudel, S., Muruganujan, A., Casagrande, J. T. & Thomas, P. D. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44, D336–342, https://doi.org/10.1093/nar/gkv1194 (2016).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2021).
Pandurangan, A. P., Stahlhacke, J., Oates, M. E., Smithers, B. & Gough, J. The SUPERFAMILY 2.0 database: a significant proteome update and a new webserver. Nucleic Acids Res 47, D490–D494, https://doi.org/10.1093/nar/gky1130 (2019).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. in Gene Prediction: Methods and Protocols (ed. Kollmar, M.) 1–14. https://doi.org/10.1007/978-1-4939-9173-0_1 (Springer New York, New York, NY, 2019).
Fan, Z. & Zhang, Z. S. A chromosomal-level genome assembly of Araneus marmoreus Schenkel, 1953 (Araneae: Araneidae) and its raw sequencing data of illumina, Pacbio and Hi-C. Science Data Bank. https://doi.org/10.57760/sciencedb.19518 (2025).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_050042785.1 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP575255 (2025).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Acknowledgements
This study was supported by grants from the Special Investigation and Classification of Invertebrates from Yintiaoling Nature Reserve (CQS24C00333). This research is also supported by the Science Foundation of School of Life Sciences SWU (20232008071901 and 20212020110501).
Author information
Authors and Affiliations
Contributions
Z.F. and Z.Z. contributed to the research design. L.W. collected the samples. L.W., L.X. T.R., L.C., J.X. and Z.F. analyzed the data. L.W., Z.F. and Z.Z. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, LY., Xiao, L., Ren, TY. et al. A chromosomal-level genome assembly of Araneus marmoreus Schenkel, 1953 (Araneae: Araneidae). Sci Data 12, 859 (2025). https://doi.org/10.1038/s41597-025-05215-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05215-1
