
Abstract
Rock carp (Procypris rabaudi) is an endemic fish species distributed in upper reaches of the Yangtze River. Due to anthropogenic influences, such as hydropower development, over-fishing and habitat destruction, the wild populations of the species have dramatically declined in recent decades. To inform effective conservation strategies, it is essential to elucidate the species’ adaptive mechanisms through genomic analyses. The present study reported a high-quality genome assembly at the chromosome level of P. rabaudi. The final genome assembly was 1.66 Gb in length, with a contig N50 of 24.95 Mb and a scaffold N50 of 32.60 Mb, and was anchored to 50 chromosomes. The assembled genome achieved a BUSCO completeness score of 97.8%, with 45,784 protein-coding genes identified, of which 94.78% were functionally annotated in at least one protein database. Additionally, 1,305 gene families were found to be expanded, while 14,463 gene families showed contraction. The high-quality reference genome of P. rabaudi served as a valuable genetic resource, providing a foundation for the conservation of wild populations and the exploration of evolutionary mechanisms.
Similar content being viewed by others
Background & Summary
The rock carp (Procypris rabaudi), is an endemic fish species distributed in the upper Yangtze River drainage, including the mainstream and its tributaries1. The species particularly inhabits environments with deep, clear, slow-moving waters and abundant rocky substrates1. Based on this habitat, P. rabaudi exhibits an omnivorous feeding behavior, with a diet primarily consisting of benthic fauna, including aquatic insects and other invertebrates, and secondarily incorporating plant detritus and phytoplankton. In terms of reproductive biology, P. rabaudi reaches sexual maturity at the age of 3 or 4 years. It selects rocky-bottomed areas in the Yangtze River and its tributaries as spawning grounds from February to April, a period that coincides with favorable water temperatures2. The yellowish ova are typically attached to submerged rocks. Procypris rabaudi is valued for its palatable flesh, making it an important commercial fish species. It has been successfully artificially propagated and bred to date. Moreover, maintaining stable populations of wild P. rabaudi is crucial for preserving biodiversity and ecological balance in the upper Yangtze River ecosystem.
However, in recent decades, the natural populations of P. rabaudi have experienced a significant decline. This decline is primarily due to anthropogenic impacts within the Yangtze River watershed, including overfishing, hydropower development, and water pollution3. As a result, it was classified as a second-class state-protected wild animal in the National Key Wild Animal List by National Forestry and Grassland Administration (2021). In response to the urgent need for conservation, researchers have analyzed the genetic diversity and historical population dynamics of the wild P. rabaudi populations using microsatellite markers and mitochondrial DNA sequences4,5. Additionally, the origins of its allotetraploidization events have been elucidated through comparative genomics and related methods6. Numerous studies have shown that P. rabaudi is polyploid species7,8, and this information is crucial for understanding the genome structure and validating the data in this study. Efforts have also focused on sex identification methods and embryonic development to optimize artificial propagation techniques9.
Sequencing and assembling reference genomes are essential tools for identifying and conserving biological diversity, especially for rare species. Reference genomes provide the foundational data necessary for dissecting the evolution of genetic elements and phenotypic traits, elucidating phylogenetic relationships, interpreting historical demographic events, assessing population genetic structures, evaluating the impacts of inbreeding and outbreeding, and predicting the capacity for demographic recovery10. Also, high-fidelity genomic data are pivotal for realizing in-depth analyses of the genome’s specific functions and regulatory mechanisms, an aspect that is currently deficient in P. rabaudi. Such knowledge is instrumental for comprehending the interplay between phenotypic expressions and genomic constitution, including morphological traits and behavioral patterns. Genomic data are essential for understanding population dynamics, uncovering mechanisms of environmental adaptation, and identifying the causes of its endangered status. Such understanding empowers researchers to concentrate on the species-specific population genetic characteristics and environmental adaptive traits, thereby informing the development of conservation strategies tailored to the unique ecological and genetic requirements of P. rabaudi. In the context of artificial breeding programs, genomic information is conducive to the design of efficacious breeding schemes that circumvent the genetic deterioration associated with inbreeding, thereby ensuring the preservation of the population’s genetic integrity. Moreover, sharing the high-quality P. rabaudi genome sequence in public databases will enhance its value for broader scientific research and conservation efforts. Thus, a high-quality genome assembly of the P. rabaudi represents a critically needed resource at this juncture.
In this study, we generated a high-quality de novo chromosome-level genome assembly of P. rabaudi using Illumina, PacBio, and chromosome conformation capture (Hi-C) sequencing technologies. The genome assembly length was 1.66 Gb, with a scaffold N50 of 32.60 Mb and a contig N50 of 24.95 Mb containing 50 chromosomes. Importantly, P. rabaudi is a polyploid species, and this chromosome-level assembly provided valuable insights into its genomic structure. We identified 803.56 Mb of repetitive elements, representing 48.38% of the genome, and annotated 43,359 protein-coding genes. This chromosome-level genome assembly provided a crucial foundation for exploring the phylogenetic relationships of this endangered and protected species. It also serves as an invaluable resource for future research into the molecular biology of P. rabaudi. This genomic resource will significantly facilitate conservation efforts and enhance our understanding of its evolutionary history.
Methods
Samples collection and whole-genome sequencing
An artificially bred adult female P. rabaudi (three years old) was collected from the Fish Reproduction Station of Ertan-Tongzilin, Panzhihua City, Sichuan Province, China. The fish was anesthetized with tricaine methanesulfonate (MS-222), then the muscle tissues were sampled and stored at −80 °C for subsequent analysis. Genomic DNA was subsequently isolated using the QIAGEN DNeasy Blood & Tissue Kit (QIAGEN, Shanghai, China). Total RNA was extracted from muscle tissues using Trizol reagent (Invitrogen, Frederick, MD, USA), and purified using a Qiagen RNeasy mini kit (Qiagen, Germantown, MD, USA). All animal experiments were conducted under the ethical guidelines of the Animal Care and Use Committee of College of Life Sciences, Sichuan University.
Subsequently, HiFi long-read sequencing was performed on the PacBio Sequel II platform (Pacific Biosciences), generating 80.36 Gb of high-fidelity sequences with an average read length of 17.36 kb, providing 48× coverage of the genome (Table 1). Additionally, the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) was used for both cDNA and genomic DNA sequencing, supporting transcriptome analysis for gene annotation and refinement of the long-read sequences, respectively.
For PacBio library construction, DNA was sheared using a Covaris g-Tube, and a SMRTbell library was prepared using the SMRTbell Express Template Preparation Kit (PacBio) according to the manufacturer’s instructions. RNA-seq libraries were constructed and sequenced on the Illumina NovaSeq6000, generating 6.23 Gb of RNA-Seq reads (Table 1). To improve scaffold anchoring, Hi-C libraries were prepared using the NEBNext® Ultra™ RNA Library Prep Kit (NEB, UK) and sequenced on the Illumina NovaSeq6000, generating 189.80 Gb of data and 114 × genomic coverage (Table 1).
Genome survey and de novo assembly
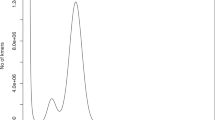
K-mer analysis was performed to estimate genome size and heterozygosity as well as repetitive sequence information based on HiFi data using the software Jellyfish v 2.3.011, showing a clear main peak when the K-mer depth was about 49. The P. rabaudi genome was estimated to be 1.64 Gb in size (Fig. 1).

Frequency distribution of K-mer depth and number of K-mer species in P. rabaudi.
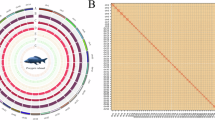
The preliminary genomes were assembled using hifiasm v0.19.6-r59512 with 80.36 Gb of cleaned HiFi reads and default parameters, resulting in a genome assembly of 1.66 Gb in size, with a contig N50 of 24.95 Mb. Hi-C DNA reads generated from Illumina sequencing were mapped to the preliminary genome using the Burrows-Wheeler Aligner (BWA)13. Contigs were anchored to 306 scaffolds based on interaction affinities between reciprocal sequence pairs. Hi-C contact matrices were constructed using Juicer software14. Subsequent correction of assembly errors and contig scaffolding were carried out using the 3D de novo assembly (3D-DNA) pipeline15. The final assembly was manually adjusted using the Juicebox Assembly Tool15 to ensure scaffolds within the same linkage group exhibited the expected Hi-C linkage pattern. After scaffolding, a total of 50 chromosomes were delineated (Fig. 2). The final genome assembly spanned 1.66 Gb, with a contig N50 of 24.95 Mb and contained 1,763 gaps (Table 2). The chromosome lengths ranged up to a maximum of 485.11 Mb (Table 3). The chromosome-level genome was visualized using Circos software16, providing a graphical representation of the genome architecture (Fig. 3).

Genome-wide Hi-C interactive heatmap of 50 pseudo-chromosomes of P. rabaudi.

Circos plot of genomic characteristics and annotation of the P. rabaudi genome assembly (1 Mb window size). From outer to inner ring: chromosome (Mb), gene density, GC content, and transposable element density. The higher the content density, the darker the color in the heatmap.
Annotation of repetitive sequences and genes
Transposable elements (TEs) in the P. rabaudi genome were identified through a combination of homology-based and de novo prediction methods. First, a de novo repeat library was constructed using Repeatmodeler17 and merged with the Repbase library. Subsequently, TEs in the genome were identified and classified based on the merged library using Repeatmasker18. The analysis showed that the repetitive TEs accounted for about 48.38% of P. rabaudi total genome, with repeated DNA element being the most abundant (~26.20%), among which long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs) and long terminal repeats (LTRs) accounted for 5.34%, 0.42% and 4.55%, respectively (Table 4).
Predictions for protein-coding genes (PCGs) were based on ab initio computed gene predictions, homology predictions, and transcript predictions. Ab initio gene prediction was performed by Augustus19 and GlimmerHMM20. For homology prediction, protein sequences of zebrafish (Danio rerio) and spotted gar (Lepisosteus oculatus) were obtained from the Ensembl database21 and aligned with P. rabaudi genome by GeneWise22. For transcriptome annotation, RNA-seq reads were aligned to P. rabaudi genome by HISAT223, and genome prediction was performed using the PASA pipeline24. All evidences above were integrated by EVidenceModeler25 to generate a consensus and non-redundant gene set. In total, we identified 43,394 predicted PCGs within the genome, representing 94.78% of all genes (Table 5).
Data Records
All raw genomic sequencing data have been submitted to the NCBI under the BioProject accession number PRJNA114120126. Raw reads are available in the Sequence Reads Archive (SRA) under accession numbers SRR3344467527 and SRR3344467428. The genome assembly data are accessible via GenBank under the Whole Genome Shotgun (WGS) project accession number JBMMUR00000000029. The annotation files of the P. rabaudi genome are available in the figshare database: https://doi.org/10.6084/m9.figshare.28451315.
Technical Validation
Evaluation of the genome assembly
P. rabaudi is a tetraploid species with a chromosome number of 2n = 100, as confirmed by previous studies6,7,8 and supported by our genomic analysis. The tetraploid nature of P. rabaudi has important implications for the assembly and annotation of its genome, as it may result in duplicated regions and increased genomic complexity. The completeness of gene coverage was assessed using the Benchmark Universal Single Copy Homologue (BUSCO) analysis30, querying the BUSCO database (actinopterygii_ODB10). This analysis evaluated the completeness, accuracy, and continuity of the genome assembly. The results showed that the integrity of the P. rabaudi genome reached 97.8%, which was higher than genome integrity (89.4%) of Gymnocypris przewalskii, a tetraploid Cyprinid fish as well31. The percentage of missing BUSCOs in the P. rabaudi genome (1.81%) was lower than expected, suggesting that complex genomic regions were accurately assembled (Table 5). We identified 43,394 protein-coding genes (PCGs) in the P. rabaudi genome through multiple annotation methods, representing 95.06% of all genes (Table 6, Fig. 4). BUSCO analysis indicated that the predicted gene integrity was 94.78% (Table 5).

Venn diagram displaying the matches of genes of P. rabaudi in four public protein databases.
Code availability
No specific code was developed in this work. The parameters of all commands and pipelines used for data processing are described in the Methods section. If no detailed parameters are mentioned for a software, the default parameters were used, as suggested by the developer.
References
Ding, R. The Fishes of Sichuan, China. Sichuan Publishing House of Science and Technology, Chengdu, Sichuan, China (1994).
Cai, Y. Z. et al. Preliminary studies on the biological characteristics of Procypris rabaudi (Tchang). J. Hydroecol. 23, 17–19 (2003).
Yang, L., Mayden, R. L. & Cai, Y. Threatened fishes of the world: Procypris rabaudi (Tchang, 1930) (Cyprinidae). Environ. Biol. Fish 84, 275–276 (2009).
He, W. et al. Three Mitochondrial Markers Reveal Genetic Diversity and Structure of Rock Carp (Procypris rabaudi) Endemic to the Upper Yangtze: Implications for Pre-release Genetic Assessment. Front. Mar. Sci. 9, 939745 (2022).
Zhang, X. et al. Isolation and characterization of nine polymorphic microsatellite loci in the rock carp, Procypris rabaudi (Tchang). Mol. Ecol. Resour. 8, 123–125 (2008).
Xu, M. R. et al. Maternal dominance contributes to subgenome differentiation in allopolyploid fishes. Nat. Commun. 14, 8357 (2023).
Xu, B. et al. Karyotype analysis of Procypris rabaudi (Tchang). J. Northwest A & F Univ. (Nat. Sci. Ed.) (2014).
Leggatt, R. A. & Iwama, G. K. Occurrence of polyploidy in the fishes. Rev. Fish Biol. Fish. 13, 237–246 (2003).
Tuo, Y. Summary of reproductive biology of rock carp. Anhui Agric. Sci. Bull. 14, 141–142 (2008).
Morin, P. A. et al. Reference genome and demographic history of the most endangered marine mammal, the vaquita. Mol. Ecol. Resour. 21, 1008–1020 (2021).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 4, 4.10.11–14.10.14 (2009).
Stanke, M. et al. AUGUSTUS:: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM:: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Flicek, P. et al. Ensembl 2024. Nucleic Acids Res. 52, D1–D7, https://www.ensembl.org (2024).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907 (2019).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
NCBI Bioproject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1141201 (2024).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR33444674 (2025).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR33444675 (2025).
NCBI GenBank https://www.ncbi.nlm.nih.gov/nuccore/JBMMUR000000000 (2025).
Manni, M. et al. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 1, e323 (2021).
Tian, F. et al. Chromosome-level genome of Tibetan naked carp (Gymnocypris przewalskii) provides insights into Tibetan highland adaptation. DNA Res. 29, 1–11 (2022).
Acknowledgements
We would like to thank the Fish Reproduction Station of Ertan-Tongzilin for providing experimental fish, and the financial support by the Yalong River Hydropower Development Company, Ltd. (No. YLTZ-TZA-ZD2023554).
Author information
Authors and Affiliations
Contributions
Zhongyi Wang was responsible for sample collection, data compilation, figure creation, and manuscript writing. Miling Ran contributed to the illustrating figures. Xiaoshuai Liu provided experimental samples. Chuang Zhou conducted data processing and analysis. Zhaobin Song contributed the conceptualization and revision of the manuscript for this study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Z., Ran, M., Liu, X. et al. Chromosome-level genome assembly of rock carp (Procypris rabaudi). Sci Data 12, 914 (2025). https://doi.org/10.1038/s41597-025-05248-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05248-6
This article is cited by
-
A chromosome-level reference genome of the surf parrotfish (Scarus rivulatus)
Scientific Data (2025)
