
Abstract
The Andean killifish Orestias ascotanensis inhabits the high-altitude Ascotán Salt Pan, an environment with variable salinity, high UV exposure, low oxygen, and extreme daily temperature fluctuations. These conditions make it an excellent model for studying high-altitude fish biology. However, the transcriptomic responses of O. ascotanensis to seasonal acclimation remain unexplored. To investigate seasonal and tissue-specific transcriptomic profiles, RNA-seq was performed on 42 libraries from gills, skin, and muscle tissues of 14 individuals collected in summer (n = 7) and winter (n = 7). Each library had a median of 105 million reads. Principal component analysis revealed strong tissue-specific expression, and seasonal differential expression analyses identified significant transcriptomic changes within each tissue. Additionally, a bioinformatics pipeline identified 10,365 high-confidence long non-coding RNAs (lncRNAs), predicted by at least three computational tools. Compared to protein-coding genes, lncRNAs exhibited higher tissue specificity, with a predominance of monoexonic structures and shorter exon lengths. This dataset provides the first comprehensive view of seasonal mRNA and lncRNA expression in O. ascotanensis tissues.
Similar content being viewed by others
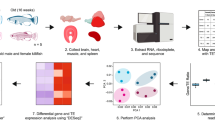

Background & Summary
Orestias ascotanensis is a killifish endemic to the springs of the Ascotán Salt Pan on the Chilean Andean Plateau, located at an altitude over 3,600 meters above sea level (m.a.s.l.). This environment is marked by challenging conditions for animal life, including extreme diurnal temperature fluctuations, low humidity, reduced oxygen partial pressure, and increased exposure to UV radiation1. Despite these challenges, various organisms, including snails2,3, frogs4, fishes5, birds6, and camelids7, have managed to thrive in this harsh environment. While genomics and transcriptomic technologies have shed light on molecular adaptations to high-altitude living, primarily from studies on the Tibetan Plateau8,9,10,11,12,13,14,15,16, the unique adaptations of Andean species remain less explored. The genus Orestias, encompassing species from various aquatic systems across the Andean basins in Peru, Bolivia, and Chile, exhibits remarkable adaptability to both saltwater and freshwater environments, confronting low oxygen concentrations, high UV radiation, and variable heavy metal and salt concentrations derived from South American summer monsoons17. Embryonic analysis of O. ascotanensis revealed early pigmentation development and increased blood cell production, suggesting adaptations to UV radiation and low oxygen pressure18. The recent sequencing of O. ascotanensis genome highlighted genes under positive selection to DNA repair, indicating a mechanism for maintaining genome integrity under the high UV exposure19. These unique features position O. ascotanensis as a valuable model for understanding fish adaptations to high-altitude environments.
Fish biology is intricately tied to seasonal changes, that significantly influences their reproductive strategies to align offspring production with optimal environmental conditions. Consequently, this seasonality exerts a profound influence on various aspects of fish life, including reproductive timing, body mass variations, locomotion, and immune system performance20,21,22. In temperate aquatic ecosystems, these fluctuations are mainly driven by changes in temperature and photoperiod, which are closely interconnected and cyclical in nature. For instance, during the spring, water temperatures and light periods increase while the reverse occurs in autumn. To adapt to these environmental shifts, fish undergo compensatory responses at both cellular and molecular levels. These adaptations involve changes in the proteome23 and the transcriptome, inducing the expression of both protein-coding mRNAs and non-coding RNAs (ncRNAs)24, alterations in DNA methylation patterns25, and histone modifications26. Recent studies have also identified the role of long non-coding RNAs (lncRNAs) in modulating seasonal behavioral patterns, including migration and spawning27.
By definition, lncRNAs are RNA molecules exceeding 200 nucleotides in length, that are not translated into proteins. Their role in modulating gene expression has garnered significant attention, influencing a variety of biological processes28. LncRNAs operate through diverse mechanisms: they can function as molecular guides by directing chromatin modifying enzymes to target genes, as scaffolds by forming ribonucleoprotein complexes, or as decoys by binding to and sequestering transcription factors away from their target genes29. Additionally, lncRNAs can regulate microRNA (miRNA) activity by serving as competing endogenous RNAs (ceRNAs). By harboring miRNA-binding sites, they can effectively sequester miRNAs, preventing them from repressing their target protein-coding genes and thereby modulating gene expression30. Despite the explosion of next-generation sequencing efforts that have identified thousands of lncRNAs across a wide range of organisms, including mammals, zebrafish, insects, and nematodes, the cataloging of lncRNAs in non-model fish species remains limited. Currently, lncRNA studies in fish focus on model organisms and commercially important species, leaving lncRNAs from other fish species unexplored.
Increasing evidence demonstrates the transcriptomic responses of fish to changes in temperature, photoperiod and other abiotic factors under controlled laboratory conditions31. However, field-based studies that explore these dynamics in natural settings are notably rare. Our study addresses this gap by leveraging the Ascotán Salt Pan’s distinct seasonal variations as a “natural laboratory” to examine the molecular responses of Orestias ascotanensis. We used the pronounced temperature and photoperiod contrasts between the Ascotán Salt Pan’s summer and winter – specifically in January and July –to profile both mRNA and lncRNA transcriptomes, seeking to delineate gene expression changes across tissues tied to seasonal acclimatization. We selected gills, muscle, and skin tissues for analysis, given their documented sensitivity to environmental shifts32,33,34. This choice is further justified by the tissue-specific nature and lower sequence conservation of lncRNAs compared to protein-coding mRNAs35, suggesting that examining multiple tissues can reveal both common and unique transcriptomic responses. Our multi-tissue approach offers an in-depth look at the Andean killifish’s transcriptional adaptation to environmental changes, encompassing both protein-coding and long non-coding RNAs. In this work, we obtained and released a total of ~4.38 billion raw reads across 42 transcriptome libraries from gill, skin, and muscle tissues of Orestias ascotanensis. These libraries, representing 14 individuals sampled during summer and winter, yielded high-quality data with a median of 105 million raw reads and 92.85 million mapped reads per library, achieving an overall mapping efficiency of 88.45%. The unique ecological and physiological adaptations of O. ascotanensis to its high-altitude environment make this transcriptome an invaluable resource for understanding tissue-specific and seasonal expression patterns, contributing to the study of adaptation and potentially speciation of endemic fish in the Andean Plateau.
Methods
Ethical statements and sample collection
All procedures for the collection and handling of adult Orestias ascotanensis specimens were conducted in strict accordance with ethical guidelines of Universidad Andrés Bello, under the official permission issued by this institution (‘Resolución n° 2267’) and of the ‘Subsecretaría de Pesca y Acuicultura’, granted to Dr. Rodrigo Maldonado-Agurto. Specimens were collected from the Ascotan Salt Pan (21°29′46.5″S 68°15′26.3″W) at an elevation of 3,740 meters above sea level in July 2019 (n = 7) and January 2020 (n = 7) using a fishing net. Euthanasia was performed by administering an overdose of tricaine methanesulphonate (MS-222, 500 mg L−1; Sigma-Aldrich). Tissue samples from gills, skin, and muscle were harvested on-site. Each tissue sample was individually preserved in RNAlater (Invitrogen) immediately after dissection to stabilize and protect RNA. The samples were transported on ice to the laboratory and subsequently stored at −80 °C until RNA extraction.
RNA extraction and sequencing
Total RNA was extracted from three different tissues (gills, skin, and muscle) samples obtained from each of the 14 individuals captured during either summer (n = 7) or winter (n = 7), using TRIzol (Invitrogen) following the manufacturer’s protocol. The concentration and quality of the extracted RNA were assessed using a Qubit fluorometer (Invitrogen) and a Bioanalyzer 2100 (Agilent), determining the integrity and purity of each sample. Unstranded poly-A libraries were prepared from each tissue sample using the Illumina TruSeq RNA sample preparation kit (Illumina) according to the manufacturer’s instructions. The prepared RNA samples were sent to Quick Biology (Pasadena, CA, USA) for sequencing, where they were processed on the Illumina HiSeq 4000 platform to generate 150 bp paired-end reads. A total of 42 RNA-seq libraries (14 individuals × 3 tissues) yielded approximately 4.4 billion raw reads, with a median of 105 million reads per library. These high-quality sequencing data provided robust coverage for transcriptome profiling across tissues and seasons.
Data processing
The transcriptomic data of Orestias ascotanensis were processed to measure protein-coding gene expression and to identify long non-coding RNAs (lncRNAs). Raw sequencing reads underwent quality control using FastQC36 and trimming with TrimGalore (v0.6.6; parameters: -q 30 --retain_unpaired --phred33--illumina)37 to remove low-quality bases and adapter sequences. High-quality reads were aligned to the O. ascotanensis reference genome using HISAT238 (v2.2.1; parameters: --dta-cufflinks), and coordinate-sorted BAM files were generated using samtools (v1.10)39. Alignment performance was evaluated per sample based on mapping statistics, including the number of raw reads, mapped reads, mapping percentages, and properly paired read percentages40.
Principal component analysis
To evaluate sample similarity and clustering based on protein-coding gene expression profiles, principal component analysis (PCA) was performed after quantifying transcript abundance. Gene-level expression matrices were generated using featureCounts41, and the raw counts were normalized with DESeq 242 to correct for library size and sequencing depth. The normalized expression data were then used for PCA, which revealed distinct clustering of samples by tissue type (gill, skin, and muscle) (Fig. 1a), underscoring the robustness of tissue-specific transcriptomic profiles. This analysis also validated the consistency of biological replicates within each tissue group, ensuring the reliability of subsequent differential expression analyses. Separate PCAs were performed within each tissue to examine seasonal variation (Fig. 1b–d). Muscle tissue showed the clearest seasonal separation, while gill and skin exhibited overlapping expression patterns between summer and winter samples.

Principal component analysis (PCA) of transcriptome profiles in Orestias ascotanensis. (a) PCA of all samples, colored by tissue type (gills: red, muscle: blue, skin: green). (b) Seasonal PCA for gills (summer: light red, winter: dark red). (c) Seasonal PCA for muscle (summer: light blue, winter: dark blue). (d) Seasonal PCA for skin, showing seasonal overlap (summer: light green, winter: dark green).
Tissue-specific gene expression analysis
Coding and non-coding transcript abundance was quantified using featureCounts41. To estimate differential gene expression between tissues and seasons, we used DESeq 2 integrated into the SARTOOLs package43. To visualize tissue specific protein-coding gene expression, an UpSet plot was constructed for all mRNAs with more than 50 counts in at least one tissue (Fig. 2a). The majority of expressed genes (13,261) were detected across all three tissues, indicating shared core biological functions. Muscle showed the highest tissue specificity with 763 uniquely expressed genes, while gills and skin had a lower number of uniquely expressed genes, including 568 and 199 mRNAs, respectively. Inter-tissue overlapping expression included 209 shared genes between gills and muscle, 733 genes between muscle and skin, and 821 genes between gills and skin. Differential gene expression analysis (fold change ≥ 2, adjusted p-value < 0.05) was next used to further identify genes that can be potentially relevant in these tissues. Thus, 5,277 and 2,674 genes showed significantly higher expression in gills compared to muscle and skin, respectively, with a subset of 1,981 genes exhibiting distinctly higher expression in gills (Fig. 2b). Conversely, 5,623 and 3,788 genes showed selectively higher expression in muscle compared to gills and skin, respectively, with 3,183 genes uniquely upregulated in muscle (Fig. 2c). Skin also showed distinct expression patterns, including 3,536 and 3,872 genes that were upregulated compared to gills and muscle, respectively, with 584 genes markedly upregulated in this skin tissue (Fig. 2d).

Tissue-specific gene expression analysis in Orestias ascotanensis. (a) UpSet plot showing the number of genes uniquely expressed or shared among gills, skin, and muscle tissues. The highest bar represents genes expressed in all tissues (13,261). (b) Venn diagram displaying the number of genes upregulated in (b) gills compared to muscle and skin (1,981 genes). (c) muscle compared to gills and skin (3,183 genes) and, (d) skin compared to gills and muscle (585).
Gene ontology of tissue-enriched genes
To evaluate whether the gene expression patterns reflect the functional identity of each tissue, we performed gene ontology (GO) enrichment analysis on genes exhibiting significantly higher expression in one tissue compared to the other two using Metascape44. Since the tissues were manually dissected, this analysis aimed to determine if the observed gene expression patterns correspond to the physiological roles of each tissue. In gills, tissue-enriched genes were associated with processes such as the regulation of blood circulation and ion channel activity, which are part of the functions of gills in osmoregulation and gas exchange (Fig. 3a). For muscle tissue, enriched terms included oxidative phosphorylation and muscle contraction, highlighting the transcriptional activity of genes involved in locomotion and energy metabolism (Fig. 3b). In skin, enriched processes such as melanogenesis and pigmentation, reflecting the tissue’s protective and barrier functions (Fig. 3c). These findings suggest that the gene expression profiles accurately represent the functional identity of each tissue, despite their cellular heterogeneity, and validate the use of these profiles to infer tissue-specific roles in Orestias ascotanensis.

Gene ontology (GO) enrichment of tissue-enriched genes. Heatmaps of top 20 enriched terms across genes with enriched expression in. (a) gills. (b) muscle, and. (c) skin. The color gradient of the bars reflects the p-value, with darker colors representing lower p-values (higher significance). The heatmaps were produced using Metascape.
Seasonal differential gene expression
As previously shown by the principal component analysis, muscle tissue exhibited the highest separation in gene expression patterns between the summer and winter seasons. The other two tissues displayed a significant degree of overlapping gene expression patterns. Accordingly, the number of differentially expressed genes (DEGs) (FC ≥ 2, p-adjust ≤ 0.05) between summer and winter seasons were found to be 3, 42, and 1019 in gills, skin, and muscle, respectively. In gills and skin, all the DEGs were found as upregulated in summer, while in muscle, between summer (n = 6) and winter (n = 7), 281 (28.27%) genes were upregulated in summer, whereas 713 (71.73%) genes were found to be downregulated. For the DEG analysis, the Summer Muscle sample 4 was excluded from this analysis as it had significantly fewer mapped reads (20.8%) than the other samples (appears as an outlier in the PCA analysis) (Fig. 1c).
Our subsequent GO analyses were then based on the DEGs obtained in muscle tissue as these data showed robust gene expression patterns and an evident seasonal influence. For this analysis, we sought to input the group of up- or down- regulated genes of Orestias ascotanensis into the Metascape platform. Metascape permits selecting between different species for both input and analysis, hence allowing researchers to assess genes from one organism while leveraging the functional annotations of another specie. Because Orestias data is not yet available in this platform, we selected the vastly annotated Homo sapiens information to carry out both the input and subsequent analysis. This strategy provided us with a more comprehensive GO enrichment and pathway analysis for muscle-related genes. GO term enrichment analysis of differentially expressed genes in O. ascotanensis muscle tissue during summer revealed distinct biological processes associated with seasonal variation. Upregulated genes (Fig. 4a) were enriched for processes related to hormonal regulation (‘regulation of hormone levels’ GO:0010817), secretion (‘regulation of secretion’ GO:0051046), and immune cell activity (‘granulocyte migration’ GO:0097530). In contrast, downregulated genes (Fig. 4b) were significantly associated with cellular structural changes, including ‘epithelial cell differentiation’ (GO:0030855) and ‘actin filament-based processes’ (GO:0030029), as well as key regulatory pathways such as ‘VEGFA-VEGFR2 signaling’ (WP3888). Interestingly, within this broader physiological context, we also observed a complementary pattern in the regulation of cell proliferation-related processes, with genes linked to negative regulation of proliferation being upregulated (e.g., ‘negative regulation of cell population proliferation’ GO:0008285) and those involved in mitotic control being downregulated (e.g., ‘mitotic cell cycle’ GO:0000278). These findings suggest that seasonal variation in gene expression may be linked to broader physiological adaptations in O. ascotanensis, influencing both tissue remodeling and systemic regulatory processes.

Seasonal variation in Gene Ontology (GO) Terms in O. ascotanensis muscle tissue. Top 20 enriched terms for summer. (a) upregulated and (b) downregulated genes in O. ascotanensis muscle tissue. The color gradient of the bars reflects the p-value, with darker colors representing lower p-values (higher significance). The heatmaps were produced using Metascape.
Genome-wide Identification of lncRNAs in Tissues of Orestias ascotanensis
Coordinate-sorted BAM files generated above were used as input for transcriptome assembly via the Cufflinks pipeline45, resulting in 264,422 reconstructed transcripts from gills, muscle, and skin tissues. These transcripts underwent a series of filtering steps to exclude coding sequences and other non-lncRNA features, yielding an initial set of 17,697 lncRNA candidates (Fig. 5a). Filtering began with the removal of 51 transcripts with designation “class code”. Then, transcripts that overlap with protein coding sequences were also removed using IntersectBed. Next, the remaining 105,582 sequences were filtered for similarity with proteins within the SwissProt/Uniprot database using BLASTx, performed locally using DIAMOND46. All transcripts matching proteins in this database, as determined by the specified parameters of analysis [-e 0.01 –k 5 –matrix BLOSUM62 –gapopen 11 –gapextend 1 –more-sensitive] were removed. The subsequent analysis targeted the remaining sequences (17,697) for protein coding potential, employing four different tools: CPAT47, Transdecoder48, RNAmining49, and FEELnc50 —which independently predicted 17,392, 4,878, 16,829, and 7,624 lncRNAs, respectively. A robust intersection of these predictions produced a final high-confidence 1,580 transcripts consistently identified by all four tools (Fig. 5b) and 10,365 were predicted as lncRNA by at least three of these tools. The latter group of potential lncRNAs was then utilized in the subsequent analyses.

Workflow for the Identification and filtering of lncRNA candidates in O. ascotanensis. (a) Step-by-step process for identifying and filtering long non-coding RNA (lncRNA) candidates from the assembled transcripts of O. ascotanensis using multiple computational tools. (b) Venn diagram showing the overlap among candidate lncRNAs filtered by FEELnc, RNAmining, CPAT, and Transdecoder.
Calculation of Tau scores
The Tau (τ) index serves as an indicator of the specificity or broadness of expression for a given gene or transcript within the analyzed tissues51. A higher Tau value implies a predominantly tissue-specific expression pattern, whereas a lower Tau value indicates a gene or transcript that is expressed more broadly across multiple tissues. In our study, we employed the τ index as a measure to assess tissue specificity, calculated using normalized gene expression data obtained from featureCounts41. The τ index is calculated using the formula:
Here, \(N\) is the number of tissues and \({\hat{x}}_{i}\) is the expression profile component normalized by the maximal component value. The τ values range from 0 to 1, reflecting the spectrum of gene expression specificity: genes with τ values up to 0.25 are considered ubiquitously expressed across various tissues, those with τ values between 0.25 and 0.80 reflect intermediate expression patterns, while τ values equal to or greater than 0.8 indicate tissue-specific gene expression.
Characterization of lncRNAs
The lncRNAs identified in Orestias ascotanensis were further characterized to understand their expression patterns and structural features. Tissue specificity was assessed using the Tau (τ) index, which measures the degree of expression specificity across tissues (Fig. 6a). A higher τ value indicates tissue-specific expression, while a lower value reflects broader expression. Among the putative lncRNAs, 21.9% exhibited tissue-specific expression (τ ≥ 0.80), compared to 15.9% of mRNAs. In contrast, 13.5% of the lncRNAs were ubiquitously expressed (τ ≤ 0.25), compared to 22% of mRNAs, highlighting the more specialized roles of lncRNAs. Structural comparisons revealed that lncRNAs generally differed from mRNAs in key features. Most lncRNAs were monoexonic (Fig. 6b), whereas mRNAs predominantly had multiple exons. Additionally, lncRNAs exhibited shorter exon lengths compared to mRNAs (Fig. 6c). These differences underscore the distinct characteristics of lncRNAs and their potential regulatory roles, which are often linked to their tissue-specific expression and unique genomic architecture. Interestingly, differential expression analysis revealed that both skin and muscle tissues exhibit differentially expressed lncRNAs, while no differentially expressed lncRNAs were found in gills (Table 1). In skin, it was determined that 2 of the lncRNAs are downregulated and 18 of them upregulated in the summer whereas in muscle, 128 lncRNAs were found downregulated and 49 upregulated in this summer season.

Characterization of O. ascotanensis lncRNAs. (a) Density distribution of tau scores, (b) frequency distribution of the number of exons, and (c) density distribution of exon lengths for mRNAs and lncRNAs.
Data Records
Gene expression profiles were deposited at the Gene Expression Omnibus (GEO) database under accession number GSE29410552. Raw sequencing data are available through the Sequence Read Archive (SRA) under study accession SRP55338353. Processed data, including mapping statistics and genome annotation files (reference genome, GTF annotation, and functional gene annotations), are publicly available on Figshare40.
Technical Validation
RNA integrity and library preparation
RNA integrity was evaluated using a Qubit fluorometer (Invitrogen) and a Bioanalyzer 2100 (Agilent Technologies, RNA 6000 Nano Kit), ensuring all RNA samples had RNA Integrity Numbers (RIN) ≥ 7. High-quality RNA was used to construct 42 paired-end unstranded poly-A libraries with the Illumina TruSeq RNA Sample Preparation Kit. Sequencing was performed on the Illumina HiSeq 4000 platform, generating 150 bp paired-end reads.
Quality filtering of sequencing reads
Raw sequencing reads underwent quality control with FastQC, confirming their quality met required thresholds. Adapter sequences and low-quality bases were trimmed using TrimGalore (v0.6.6; parameters: -q 30 --retain_unpaired --phred33 --illumina). Post-filtering, a total of 4.4 billion high-quality paired-end reads were retained across the 42 RNA-seq libraries, with a median of 105 million reads per library.
Read mapping and transcript assembly
Filtered reads were aligned to the O. ascotanensis reference genome using HISAT2 (v2.2.1; parameters: --dta-cufflinks), achieving a median mapping rate of 88.45%40. Coordinate-sorted BAM files were generated for downstream analyses.
Sample clustering and reproducibility
Principal component analysis (PCA) was performed to validate transcriptomic consistency across tissues and seasons. PCA of all samples revealed distinct clustering by tissue type (gill, skin, and muscle), underscoring the reproducibility of tissue-specific expression patterns (Fig. 1a).
Code availability
No specific code or script was used in this work. All commands used for data processing were executed with default settings, except in cases where specific options or parameters were required, as detailed in the Methods section.
References
Cabrol, N. A. et al. The High-Lakes Project. J Geophys Res Biogeosci 114 (2009).
Collado, G. A. & Méndez, M. A. Microgeographic differentiation among closely related species of Biomphalaria (Gastropoda: Planorbidae) from the Andean Altiplano. Zool J Linn Soc https://doi.org/10.1111/zoj.12073 (2013).
Collado, G. A., Valladares, M. A. & Méndez, M. A. A new species of Heleobia (Caenogastropoda: Cochliopidae) from the Chilean Altiplano. Zootaxa Preprint at https://doi.org/10.11646/zootaxa.4137.2.8 (2016).
Sáez, P. A. et al. A new endemic lineage of the Andean frog genus Telmatobius (Anura, Telmatobiidae) from the western slopes of the central Andes. Zool J Linn Soc https://doi.org/10.1111/zoj.12152 (2014).
Parenti, L. A taxonomic revision of the Andean killifish genus Orestias (Cyprinodontiformes, Cyprinodontidae). Bull Am Mus Nat Hist (1984).
Valqui, M., Caziani, S. M., Rocha-O., O. & Rodriguez-R., E. Abundance and distribution of the South American Altiplano Flamingos. Waterbirds https://doi.org/10.2307/1522154 (2000).
Wheeler, J. C. Evolution and present situation of the South American camelidae. Biological Journal of the Linnean Society Preprint at https://doi.org/10.1016/0024-4066(95)90021-7 (1995).
Qiu, Q. et al. The yak genome and adaptation to life at high altitude. Nat Genet https://doi.org/10.1038/ng.2343 (2012).
Yu, L. et al. Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nat Genet https://doi.org/10.1038/ng.3615 (2016).
Zhang, Q. et al. Genome resequencing identifies unique adaptations of Tibetan chickens to hypoxia and high-dose ultraviolet radiation in high-altitude environments. Genome Biol Evol https://doi.org/10.1093/gbe/evw032 (2016).
Hao, Y. et al. Comparative transcriptomics of 3 high-altitude passerine birds and their low-altitude relatives. Proc Natl Acad Sci USA https://doi.org/10.1073/pnas.1819657116 (2019).
Yang, W., Qi, Y. & Fu, J. Exploring the genetic basis of adaptation to high elevations in reptiles: A comparative transcriptome analysis of two toad-headed agamas (genus Phrynocephalus). PLoS One https://doi.org/10.1371/journal.pone.0112218 (2014).
Li, J. T. et al. Comparative genomic investigation of high-elevation adaptation in ectothermic snakes. Proc Natl Acad Sci USA https://doi.org/10.1073/pnas.1805348115 (2018).
Yang, L., Wang, Y., Zhang, Z. & He, S. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus. Genome Biol Evol https://doi.org/10.1093/gbe/evu279 (2014).
Tong, C., Tian, F., Zhang, C. & Zhao, K. The microRNA repertoire of Tibetan naked carp Gymnocypris przewalskii: A case study in Schizothoracinae fish on the Tibetan Plateau. PLoS One https://doi.org/10.1371/journal.pone.0174534 (2017).
Yang, L., Wang, Y., Sun, N., Chen, J. & He, S. Genomic and functional evidence reveals convergent evolution in fishes on the Tibetan Plateau. Mol Ecol 30 (2021).
Cruz-Jofré, F. et al. Geographical isolation and genetic differentiation: The case of Orestias ascotanensis (Teleostei: Cyprinodontidae), an Andean killifish inhabiting a highland salt pan. Biological Journal of the Linnean Society https://doi.org/10.1111/bij.12704 (2016).
Macaya, C., Lam, N. & Vila, I. Embryological development of the high-altitude killifish Orestias ascotanensis Parenti 1984 (Teleostei: Cyprinodontidae). Environ Biol Fishes https://doi.org/10.1007/s10641-019-00859-6 (2019).
Di Genova, A. et al. Genome sequencing and transcriptomic analysis of the Andean killifish Orestias ascotanensis reveals adaptation to high-altitude aquatic life. Genomics 114, 305–315 (2022).
Bromage, N., Porter, M. & Randall, C. The environmental regulation of maturation in farmed finfish with special reference to the role of photoperiod and melatonin. Aquaculture 197 (2001).
Bowden, T. J., Thompson, K. D., Morgan, A. L., Gratacap, R. M. L. & Nikoskelainen, S. Seasonal variation and the immune response: A fish perspective. Fish Shellfish Immunol 22 (2007).
Al-Emran, M., Zahangir, M. M., Badruzzaman, M. & Shahjahan, M. Influences of photoperiod on growth and reproduction of farmed fishes - prospects in aquaculture. Aquaculture Reports vol. 35 Preprint at https://doi.org/10.1016/j.aqrep.2024.101978 (2024).
Dietrich, M. A. et al. Acclimation to cold and warm temperatures is associated with differential expression of male carp blood proteins involved in acute phase and stress responses, and lipid metabolism. Fish Shellfish Immunol 76 (2018).
Qiang, J. et al. The expression profiles of miRNA-mRNA of early response in genetically improved farmed tilapia (Oreochromis niloticus) liver by acute heat stress. Sci Rep 7 (2017).
Navarro-Martín, L. et al. DNA methylation of the gonadal aromatase (cyp19a) promoter is involved in temperature-dependent sex ratio shifts in the European sea bass. PLoS Genet 7 (2011).
Pinto, R. et al. Seasonal environmental changes regulate the expression of the histone variant macroH2A in an eurythermal fish. FEBS Lett 579 (2005).
Nakayama, T. et al. Seasonal regulation of the lncRNA LDAIR modulates self-protective behaviours during the breeding season. Nat Ecol Evol https://doi.org/10.1038/s41559-019-0866-6 (2019).
Geisler, S. & Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nature Reviews Molecular Cell Biology vol. 14 Preprint at https://doi.org/10.1038/nrm3679 (2013).
Wang, K. C. & Chang, H. Y. Molecular Mechanisms of Long Noncoding RNAs. Molecular Cell vol. 43 Preprint at https://doi.org/10.1016/j.molcel.2011.08.018 (2011).
Poliseno, L. et al. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465 (2010).
Oomen, R. A. & Hutchings, J. A. Transcriptomic responses to environmental change in fishes: Insights from RNA sequencing. FACETS 2 (2017).
Fuentes, E. N., Zuloaga, R., Valdes, J. A., Molina, A. & Alvarez, M. Skeletal muscle plasticity induced by seasonal acclimatization involves IGF1 signaling: Implications in ribosomal biogenesis and protein synthesis. Comp Biochem Physiol B Biochem Mol Biol 176 (2014).
Nilsson, G. E., Dymowska, A. & Stecyk, J. A. W. New insights into the plasticity of gill structure. Respiratory Physiology and Neurobiology vol. 184 Preprint at https://doi.org/10.1016/j.resp.2012.07.012 (2012).
Hu, P. et al. Transcriptome comparison reveals a genetic network regulating the lower temperature limit in fish. Sci Rep 6 (2016).
Mercer, T. R., Dinger, M. E. & Mattick, J. S. Long non-coding RNAs: Insights into functions. Nature Reviews Genetics vol. 10 Preprint at https://doi.org/10.1038/nrg2521 (2009).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. Preprint at (2010).
Felix Krueger. TrimGalore. https://github.com/FelixKrueger/TrimGalore#readme (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Maldonado, R., Acevedo, E., Maracaja-Coutinho, V. & Montecino, M. Seasonal Transcriptome Profile Across Three Different Tissues of the Andean Killifish Orestias ascotanensis. Figshare https://doi.org/10.6084/m9.figshare.28771088.v2 (2025).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (2014).
Anders, S. Analysing RNA-Seq data with the DESeq package. R manual (2012).
Varet, H., Brillet-Guéguen, L., Coppée, J. Y. & Dillies, M. A. SARTools: A DESeq 2- and edgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One 11 (2016).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1–10 (2019).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc https://doi.org/10.1038/nprot.2012.016 (2012).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods vol. 12 Preprint at https://doi.org/10.1038/nmeth.3176 (2014).
Wang, L. et al. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res 41 (2013).
Haas, B. & Papanicolaou, A. TransDecoder. https://github.com/TransDecoder/TransDecoder (2017).
Maracaja-Coutinho, V. et al. RNAmining: A machine learning stand-alone and web server tool for RNA coding potential prediction. F1000Res 10 (2021).
Wucher, V. et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res 45 (2017).
Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21 (2005).
Maldonado, R. & Montecino, M. https://identifiers.org/geo/GSE294105. NCBI GEO (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP553383 (2024).
Acknowledgements
This work was supported by grants from ANID-FONDECYT 1211026 (MM); Millennium Institute Center for Genome Regulation ICN2021_044 (MM); Millennium Nucleus of Neuroepigenetics and Plasticity NCN2023_032 (MM); FONDECYT de postdoctorado 3190720 and FONDECYT de Iniciación 11220698 (RMA). The authors thank Dr. Miguel Allende (Universidad de Chile) for his assistance during the initial stages of this study.
Author information
Authors and Affiliations
Contributions
R.M.A. designed the experiments, collected the samples, analyzed the data and wrote the manuscript. E.A. extracted the R.N.A. and prepared the sequencing libraries. V.M. participated in the data analysis and manuscript editing. M.M. participated in experimental design, data analysis and in the preparation of the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Maldonado-Agurto, R., Acevedo, E., Maracaja-Coutinho, V. et al. Seasonal transcriptome profile across three different tissues of the Andean Killifish Orestias ascotanensis. Sci Data 12, 976 (2025). https://doi.org/10.1038/s41597-025-05345-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05345-6
