
Abstract
Litter size is a critical economic trait in the sheep industry. Like many other breeds, Duolang sheep typically produce one lamb per ewe per lambing. To date, genetic studies of this trait in that breed have largely relied on candidate gene approaches. To expand the genomic resources for this breed, we sequenced 297 genomes, generating approximately 10.52 trillion bases with an average coverage of 13.35X. High-quality alignments with a mapping rate exceeding 99% enabled the identification of 43,968,128 SNPs and 6,504,047 InDels. This dataset provides a valuable resource for identifying genetic variants associated with litter size through genome-wide association studies (GWAS) and lays the foundation for future genetic improvement efforts through genomic selection in Duolang sheep. Beyond trait mapping, the dataset also supports broader applications, including analyses of genetic diversity, phylogenetic relationships, population history, adaptive introgression, and breed-specific characteristics. Additionally, the moderate-coverage WGS data are suitable for structural variant (SV) detection and downstream analyses such as association mapping and the identification of SVs underlying phenotypic traits.
Similar content being viewed by others

Background & Summary
Litter size, defined as the number of lambs born per ewe per lambing, is a key economic trait in the sheep industry, which supplies dairy, meat, or wool for human consumption. According to bioeconomic models, increasing litter size from one to two lambs per ewe has been estimated to yield an economic gain of 20 Euros in dairy sheep populations1 and up to 690 Euros in non-dairy sheep2. Consequently, improving litter size has become a central objective in sheep breeding programs, with implications for agricultural productivity, rural development, and broader socio-economic benefits.
Among the more than 800 well-defined sheep breeds developed since domestication3, only a small number consistently produce two or more lambs per lambing. Notable high-fecundity breeds include Booroola Merino, Hu, and Cambridge sheep, in which major genes such as BMPR1B4, BMP155, and GDF96 have been identified as key regulators of prolificacy. Despite these advances, such major-effect variants are rare, and most sheep breeds still exhibit single-lamb litter sizes due to either polygenic control or insufficient selection pressure on litter size.
Duolang sheep is one such breed that generally produces one lamb per ewe per lambing. It is mainly distributed in Maigaiti, Bachu, Yupuhu, and Shache counties of Xinjiang Province, where it has evolved specific adaptations to arid desert conditions. This adaptation makes the breed an important genetic resource not only for local production systems but also for research into environmental resilience. Although some phenotypic traits, such as coat color7, or meat quality8, have been sporadically reported, increasing litter size remains a key breeding goal. For example, an earlier study identified potential associations between FSHR polymorphisms and litter size in Duolang sheep9. However, despite many candidate gene and SNP-based association studies10,11, most results have yielded weak or inconsistent associations, likely due to limited statistical power, small effect sizes, or reliance on single-marker tests.
With the rapid development of high-throughput sequencing technologies and reduced costs, whole-genome sequencing (WGS) has become a fundamental tool for identifying functional variants and major-effect genes. For instance, a WGS-based GWAS identified a deletion in the ABO blood group gene that influences gut microbiota composition in pigs12. In sheep, recent multi-omics studies incorporating WGS have linked variants in BMPR1B to litter size and PAPPA to lambing interval13.
In addition to its use in gene discovery for economic traits, WGS has increasingly been applied to broader questions in population and functional genomics, including the role of introgression in enhancing genetic diversity14, the identification of PDGFD as the causal gene for the fat-rumped tail characteristic15, the assessment of within- and between-breed genetic variation16, and the reconstruction of demographic history and phylogenetic relationships14,17. Additionally, WGS, whether at moderate or high coverage, facilitates the detection of structural variants (SVs) using either direct read-based methods18 or pangenome-guided approaches19, expanding the scope of genotype-phenotype association studies.
Here, we present a whole-genome sequencing dataset for 297 Duolang sheep from Xinjiang of China, each with a corresponding litter size phenotype (recorded as having either a single or double litter per lambing). This dataset provides a foundational resource for SNP-based association analyses and may also support genomic selection strategies for improving reproductive traits as the reference population expands. Beyond reproductive trait analysis, the high-quality variants identified from this dataset can also be used to assess genetic diversity, infer population history, examine phylogenetic relationships with other breeds, and explore breed-specific adaptations. Moreover, the WGS data are suitable for structural variant detection using both read-based and pangenome-guided approaches, enabling further exploration of SVs associated with breed-defining traits and litter size variation.
Methods
Sampling
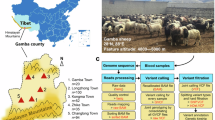
Blood samples were collected from 302 Duolang sheep at the Stock Breeding Farm in Maigaiti County, Kashi Prefecture, Xinjiang Uyghur Autonomous Region of China. The cohort consisted of 151 individuals with double-litter size records and 151 individuals with single-litter size records. Approximately 2 ml of blood was drawn from the jugular vein by a trained veterinarian using 5 ml EDTA-coated tubes. The samples were transported to the laboratory in an ice box and stored at −20 °C. An overview of the experimental workflow, including sampling, sequencing, and variant filtering steps, is shown in Fig. 1. All animal procedures were conducted in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals of China and were approved by the Animal Care Committee of Xinjiang Agricultural University.

Overview of the experimental and analytical workflow. This figure illustrates the full pipeline used in the study, including sample collection, whole-genome sequencing, read trimming, variant calling, and filtering steps. Sequencing reads were processed following standard GATK best practices for variant discovery, with the exception of applying more stringent filtering parameters in the VariantFiltration module to reduce false positives. This workflow ensures high-quality variant calls and provides a reproducible framework for downstream genomic analyses.
DNA extraction and sequencing
Genomic DNA was extracted using the standard phenol-chloroform protocol. DNA quality was assessed on 1% agarose gels, and concentrations were measured using a Qubit fluorometer (Invitrogen, Carlsbad, USA). The quantified DNA samples were shipped on dry ice to Novogene Bioinformatics Institute (Beijing, China). After DNA fragmentation, adapter ligation, and PCR amplification, 350 bp libraries were constructed for each individual. Sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina Inc., USA) using a 2 × 150 bp paired-end mode.
Read mapping and variant detection
Sequenced reads were processed using Trimmomatic v0.3920 with the parameters “LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:90” to remove low-quality bases. The filtered reads were aligned to the reference genome assembly (ARS-UI_Ramb_v3.0) using the BWA-MEM algorithm v0.7.17-r118821 with default settings. The resulting alignment files were coordinate-sorted by chromosome using Samtools v1.1722, duplicates were marked using the MarkDuplicates module in GATK v4.4.0.023, and mate-pair information was synchronized using GATK’s FixMateInformation module.
Variant calling was performed for each individual using GATK’s HaplotypeCaller module, followed by merging variant files with the CombineGVCFs module. Joint genotyping was conducted with the GenotypeGVCFs module. Hard filtering criteria were applied using the VariantFiltration module, with thresholds as follows: for SNPs, “QD < 3.0 || FS > 30.0 || SOR > 4.0 || MQ < 30.0 || MQRankSum < −10.0 || QUAL < 50.0 || ReadPosRankSum < −5.0”; and for InDels, “QD < 3.0 || FS > 100.0 || ReadPosRankSum < −10.0.” Bi-allelic variants were retained using the SelectVariants module by requiring allele count > 0 and allele frequency < 1 to exclude monomorphic and fixed sites. To further improve genotype-level accuracy, additional filtering was applied using an in-house Perl script in conjunction with BCFtools v1.1724. Specifically, we removed genotypes with genotype quality (GQ) <20 and filtered based on read depth (DP), excluding genotypes with depths <1/3× or >3× the individual’s mean whole-genome coverage. For heterozygous genotypes, we further required an allele depth ratio (AD) ≥ 0.2 to reduce the likelihood of sequencing artifacts. This filtering pipeline identified a total of 43,968,128 SNPs and 6,504,047 InDels.
Data Records
The FASTQ files for the whole-genome sequences are publicly available in the NCBI Sequence Read Archive (SRA) under accession number PRJNA1177205 (https://identifiers.org/ncbi/insdc.sra:SRP544918)25. The clean VCF file, generated using GATK, has been deposited in the European Variation Archive (EVA) under accession number PRJEB83806 (https://identifiers.org/ena.embl:PRJEB83806)26. Supplementary Table S1 provides detailed metadata linking individual sheep IDs to their corresponding NCBI BioSample IDs, sequencing alignment metrics, and litter size phenotypes. The full description of the GWAS methods and results is available in Supplementary File 1.
Technical Validation
Quality of sequencing reads
Both raw and clean sequencing reads were evaluated using Seqkit v2.6.127 and FastQC v0.11.928 to assess base counts and quality metrics. On average, each individual yielded 38.54 Gb of raw sequencing data (range: 14.04–120.13 Gb), with 99.99% and 90.32% of bases achieving Phred quality scores of 20 (base accuracy of 99%) and 30 (base accuracy of 99.9%), respectively. After quality control, an average of 35.65 Gb of clean reads per individual was retained (range: 12.96–110.60 Gb), with 100% of reads reaching Q20 and more than 90.87% reaching Q30. The median GC content across samples was 42.27% (range: 40%–44%), consistent with values reported in previous sheep genome studies29 (Fig. 2).

Quality assessment of sequencing reads across 302 Duolang sheep. This figure presents density plots summarizing key quality metrics of the sequencing data. (a) Total number of bases generated per individual; (b) Total number of reads per individual; (c) GC content distribution across individuals; (d) Proportion of reads with a mean base quality score >20; and (e) Proportion of reads with a mean base quality score >30. These results demonstrate overall high sequencing quality and consistency across samples, supporting the reliability of downstream variant detection and analysis.
Quality of alignments
Alignment quality was assessed using the bamqc command from Qualimap v2.2.130. Summary statistics are provided in Supplementary Table S1. Based on the ratio of the mean sequencing depth on the Y chromosome to the genome-wide average (approximately 0.5), five individuals were identified as male and subsequently excluded from further analyses. Across the remaining samples, the median sequencing depth was 13.05× (range: 4.96× to 44.48×), and the median mapping ratio was 99.91% (range: 98.99% to 99.97%). The median mapping quality score was 47.96 (range: 47.55 to 48.30) (Fig. 3). These metrics are consistent with those reported in previous whole-genome sequencing studies in sheep15,29, indicating high alignment quality suitable for downstream variant detection.

Alignment quality metrics across 297 Duolang ewes. This figure shows density plots summarizing key alignment statistics following read mapping to the reference genome. (a) Mapping ratio, indicating the proportion of reads successfully aligned to the genome; (b) Mean sequencing depth per individual, reflecting the average number of reads covering each genomic position; and (c) Average mapping quality score, representing the confidence in alignment accuracy. The consistently high mapping metrics across samples demonstrate reliable data quality suitable for downstream variant analysis.
Quality of variants
After the joint calling procedure, we applied GATK’s recommended ‘hard filters’ approach, a widely accepted and robust method for reducing false positives in variant calling31. To assess the accuracy of variant calling, we calculated the transition-to-transversion (Ti/Tv) ratio, a commonly used indicator of variant calling quality. The calculated Ti/Tv ratio was 2.39, which falls within the range reported in previous studies on sheep32,33, suggesting that the parameters used for variant calling and filtering were appropriately calibrated.
Functional annotation of the variants was performed using ANNOVAR v2016-02-0134. Consistent with findings from other studies in sheep17,35, the majority of variants were located in intergenic (54.39%) and intronic (39.67%) regions. Among the smaller proportion of variants found in exonic regions (2.21%), 174,533 variants resulted in amino acid changes, 3,745 caused stop codon alterations, and 35,873 led to frameshift mutations. The chromosomal distribution of variants, visualized using CMplot v4.5.136 was generally uniform except for chromosome 10 (Fig. 4), a pattern that aligns with observations in previous studies on sheep37,38.

Functional annotation and chromosomal distribution of detected variants. (a) Functional classification of SNPs and InDels based on gene structure, showing the proportion of variants located in intergenic, intronic, exonic, and other genomic regions. (b) Density plot illustrating the genomic distribution of variants across autosomes and the X chromosome. The variants are generally evenly distributed, with minor deviations observed on specific chromosomes, consistent with known genomic features in sheep.
Code availability
Data analyses were primarily performed using standard bioinformatics tools on the Linux operating system. Detailed information regarding software versions, code parameters, and in-house Perl and R scripts is available at https://github.com/915813786/Duolang-sheep-for-litter-size.
References
Wolfová, M., Wolf, J., Krupová, Z. & Margetín, M. Estimation of economic values for traits of dairy sheep: II. Model application to a production system with one lambing per year. Journal of dairy science 92, 2195–2203 (2009).
Wolfová, M., Wolf, J. & Milerski, M. Calculating economic values for growth and functional traits in non‐dairy sheep. Journal of Animal Breeding and Genetetics 126, 480–491 (2009).
Teletchea, F. in Animal Domestication Ch. Chapter 1 A Brief Overview, 137–159 (Intechopen, 2019).
Souza, C., MacDougall, C., Campbell, B., McNeilly, A. & Baird, D. The Booroola (FecB) phenotype is associated with a mutation in the bone morphogenetic receptor type 1 B (BMPR1B) gene. Journal of Endocrinology 169, R1 (2001).
Hanrahan, J. P. et al. Mutations in the genes for oocyte-derived growth factors GDF9 and BMP15 are associated with both increased ovulation rate and sterility in Cambridge and Belclare sheep (Ovis aries). Biology of reproduction 70, 900–909 (2004).
Abdoli, R., Zamani, P., Mirhoseini, S., Ghavi Hossein‐Zadeh, N. & Nadri, S. A review on prolificacy genes in sheep. Reproduction in domestic animals 51, 631–637 (2016).
Yang, G.-L. et al. Mutations in MC1R gene determine black coat color phenotype in Chinese sheep. The Scientific World Journal 2013, 675382 (2013).
Yan, X. et al. The postmortem μ-calpain activity, protein degradation and tenderness of sheep meat from Duolang and Hu breeds. International Journal of Food Science Technology 53, 904–912 (2018).
Tao, M., Li, Z., Liu, M., Ma, H. & Liu, W. Association analysis of polymorphisms in SLK, ARHGEF9, WWC2, GAB3, and FSHR genes with reproductive traits in different sheep breeds. Frontiers in Genetics 15, 1371872 (2024).
Niu, Z.-G., Wang, S., Chang, L. & Shi, H.-C. Correlation analysis between NCOA 1 gene polymorphism and fertility trait of Duolang sheep. Jiangxi Academy of Agricultural Sciences 32, 118–122 (2020).
Shi HongCai, S. H. et al. Detection of Fec B mutation and its relationship with litter size in Xinjiang Duolang sheep (Ovis aries). Journal of Agricultural Biotechnology 19, 330–334 (2011).
Yang, H. et al. ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs. Nature 606, 358–367 (2022).
Han, B. et al. Multiomics Analyses Provide New Insight into Genetic Variation of Reproductive Adaptability in Tibetan Sheep. Molecular Biology Evolution 41, msae058 (2024).
Lv, F.-H. et al. Whole-genome resequencing of worldwide wild and domestic sheep elucidates genetic diversity, introgression, and agronomically important loci. Molecular biology evolution 39, msab353 (2022).
Li, X. et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nature communications 11, 2815 (2020).
Stoffel, M. A., Johnston, S. E., Pilkington, J. G. & Pemberton, J. M. Genetic architecture and lifetime dynamics of inbreeding depression in a wild mammal. Nature communications 12, 2972 (2021).
Sun, L. et al. Resequencing reveals population structure and genetic diversity in Tibetan sheep. BMC genomics 25, 906 (2024).
Yang, J. et al. Structural variant landscapes reveal convergent signatures of evolution in sheep and goats. Genome Biology 25, 148 (2024).
Li, R. et al. A sheep pangenome reveals the spectrum of structural variations and their effects on tail phenotypes. Genome Research 33, 463–477 (2023).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303 (2010).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP544918 (2024).
European Variation Archive. https://identifiers.org/ena.embl:PRJEB83806 (2024).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PloS one 11, e0163962 (2016).
Andrews, S. FastQC: a quality control tool for high throughput sequence data, Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Zhao, H. et al. Whole-genome re-sequencing association study on yearling wool traits in Chinese fine-wool sheep. Journal of Animal Science 99, skab210 (2021).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Hemstrom, W., Grummer, J. A., Luikart, G. & Christie, M. R. Next-generation data filtering in the genomics era. Nature Reviews Genetics, 1–18 (2024).
Guo, Y. et al. Sequencing reveals population structure and selection signatures for reproductive traits in Yunnan semi-fine wool sheep (Ovis aries). Frontiers in Genetics 13, 812753 (2022).
Tian, D. et al. Genetic diversity and selection of Tibetan sheep breeds revealed by whole-genome resequencing. Animal bioscience 36, 991 (2023).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164–e164 (2010).
Zhang, W. et al. Whole-Genome Resequencing Reveals Selection Signal Related to Sheep Wool Fineness. Animals 13, 2944 (2023).
Yin, L. et al. rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics, Proteomics and Bioinformatics 19, 619–628 (2021).
Zhu, M. et al. Whole-genome resequencing of the native sheep provides insights into the microevolution and identifies genes associated with reproduction traits. BMC genomics 24, 392 (2023).
Wang, Z. et al. Genome-wide detection of CNVs and association with body weight in sheep based on 600 K SNP arrays. Frontiers in genetics 11, 558 (2020).
Acknowledgements
This project was funded by the Tianshan Talent Project of the Xinjiang Uygur Autonomous Region (2023TSYCLJ0017), the Xinjiang Agriculture Research System (XJARS-09-04) and the China Scholarship Council (201907650025). Computational resources have been provided by the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds de la Recherche Scientifique de Belgique (F.R.S.-FNRS) under Grant No. 2.5020.11 and by the Walloon Region.
Author information
Authors and Affiliations
Contributions
C.F., F.F. and W.L. conceived the research project. H.C. collected the blood sample and calving records, extracted genomic DNA, and uploaded whole genome sequences. C.F. and Q.C. did bioinformatics analysis. C.F. uploaded the clean VCF file. C.F. and Q.C. wrote the draft. W.L. acquired the funding. T.D. and F.F. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fang, C., Druet, T., Cao, H. et al. Whole genome sequences of 297 Duolang sheep for litter size. Sci Data 12, 1086 (2025). https://doi.org/10.1038/s41597-025-05448-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05448-0
