
Abstract
Meretrix is a commercially valuable bivalve genus in Asia, but only one reference genome has hindered comprehensive genetic studies and germplasm resource evaluation. In this study, we present three reference genomes of Meretrix species: Meretrix sp. MF1, Meretrix sp. MT1, and Meretrix lamarckii JML1. Meretrix sp. MF1 was assembled at the chromosome level using Nanopore sequencing and Hi-C technologies, whereas Meretrix sp. MT1 and Meretrix lamarckii were assembled as scaffold-level assemblies. The chromosome-level genome of Meretrix sp. MF1 consists of 36 contigs, including 19 chromosomes and 17 scaffolds, with a total length of 883.3 Mb and a scaffold N50 of 46.87 Mb. Notably, the genome of Meretrix sp. MF1, a putative novel species, exhibits an Average Nucleotide Identity (ANI) of 94.33% with its closest relative, Meretrix lamarckii. These genomic resources not only provide a crucial foundation for genetic research on Meretrix but also contribute to the development of effective conservation strategies for its sustainable management.
Similar content being viewed by others

Background & Summary
The genus Meretrix is a commercially significant marine bivalve widely distributed across the warm coastal waters of East and Southeast Asia1. It is particularly abundant along the southern Taiwan coastline, where it has become one of the most economically valuable species in aquaculture2. Meretrix thrives in water temperatures ranging from 25 °C to 33 °C, with significant growth slowing below 20 °C and mass mortality occurring when temperatures exceed 45 °C. Additionally, it prefers salinities between 16 and 35 ppt, with extreme fluctuations in salinity adversely affecting its survival and development3. Due to this environmental sensitivity, Meretrix aquaculture has recently suffered from slowed growth and mass mortality linked to climate change, directly contributing to the dramatic decline in production observed in Taiwan. Historically, a single hectare of culture area could yield up to 18 metric tons, but current yields have plummeted to as low as 0.6 metric tons4. Beyond environmental degradation and climate change, other contributing factors to this decline include disease outbreaks, improper aquaculture management, and genetic deterioration due to inbreeding5.
Despite the economic and ecological significance of Meretrix, genomic resources for this genus remain scarce. To date, the genome of only M. petechialis has been published6, and the morphological similarities among various Meretrix species present challenges for accurate classification and genetic studies. A high-quality reference genome is essential for understanding the genetic basis of adaptive evolution, population dynamics, and potential genetic vulnerabilities within Meretrix species. Moreover, genomic data could shed light on mechanisms underlying disease resistance, stress tolerance, and reproductive strategies, all of which are critical for the sustainable management and conservation of these species. Meretrix species are commonly found in the coastal and estuarine areas of Taiwan. However, these two habitats exhibit distinct environmental conditions. Coastal waters typically maintain higher salinity levels, ranging from 32 to 35 psu, whereas estuarine areas experience greater salinity fluctuations, potentially varying from 0.5 to 35 psu. Therefore, in this study, we collected Meretrix samples from these two contrasting environments. Meretrix sp. MF1 was specifically collected from the open coastal waters (Anping, Tainan), while Meretrix sp. MT1 was exclusively obtained from the estuarine environment (Cigu, Tainan). Our Previously study has showed that the Meretrix lamarckii clade is divided into two main distinct groups: one containing sample collect from Japan, and the other containing samples from Taiwan, suggesting that M. lamarckii from Taiwan and M. lamarckii from Japan are distinct species7. Therefore, we selected Meretrix sp. MF1, a potential novel species, for high-quality chromosome-level genome assembly. As there is no reference genome for M. lamarckii currently, M. lamarckii JML1 from Japan was also selected for genome assembly. On the other hand, Meretrix sp. MT1, MT2, and MT3, collected from the coastal waters of Taiwan, formed a distinct clade and were most closely related to M. lusoria from China. Meretrix sp. MT1 was selected for genome assembly.
In this study, we present chromosome-level genome assemblies of one Meretrix species, using a combination of Illumina short-read sequencing, Nanopore long-read sequencing, and Hi-C chromatin conformation capture technologies. For Meretrix sp. MF1, we generated a total of 51.1 Gb of Illumina data, 80.02 Gb of Nanopore data, and 46.48 Gb of Hi-C data. The final assembly yielded 19 chromosomes with a total length of approximately 883.3 Mb and a scaffold N50 of 46.87 Mb. Based on this high-quality reference genome, we successfully assembled the genomes of two additional Meretrix species, Meretrix sp. MT1 and M. lamarckii JML1. For Meretrix sp. MT1, we obtained 56.6 Gb of Illumina data and 66.79 Gb of Nanopore data, resulting in the assembly of 19 chromosomes with a total length of 944.74 Mb. Similarly, for M. lamarckii JML1, we obtained 42.6 Gb of Illumina data and 88.91 Gb of Nanopore data, resulting in the assembly of 19 chromosomes with a total length of 883.07 Mb. Meretrix sp. MF1 was historically regarded as conspecific with Meretrix lamarckii due to their indistinguishable external morphology. However, our preliminary studies based on mtDNA COI revealed distinct genetic differences between the two. To further explore the genetic relationships among these species, we conducted comparative genomic analyses and average nucleotide identity (ANI) calculations. In this study, our results further demonstrate that Meretrix sp. MF1 and M. lamarckii JML1 exhibit genomic divergence with an ANI of 94.33%. Additionally, estimated divergence times among Meretrix species inferred from metazoan orthologous genes indicated further divergence. These lines of evidence consistently support the conclusion that Meretrix sp. MF1 is a cryptic species within the genus Meretrix and should not be considered conspecific with M. lamarckii. Based on these findings, we consider Meretrix sp. MF1 to be a novel species, distinct from M. lamarckii. However, its formal taxonomic status remains pending further morphological and taxonomic investigation.
The high-quality reference genome presented in this study provides a valuable foundation for future research on Meretrix population genomics, adaptive evolution, and genetic diversity. It will also facilitate further studies on gene function, aquaculture enhancement, and sustainable aquaculture practices. Additionally, our findings highlight the importance of genomic resources in identifying cryptic species, understanding evolutionary processes, and supporting sustainable aquaculture efforts. The availability of this genomic data will empower researchers and aquaculture practitioners to develop targeted breeding programs and genetic management strategies, ultimately enhancing the resilience and productivity of Meretrix populations in the face of environmental challenges.
Methods
Sampling and nucleic acid extraction
Samples of Meretrix sp. MF1 were collected from the coastal waters of southern Taiwan (Anping, Tainan), while Meretrix sp. MT1 was obtained from the estuarine region of southern Taiwan (Cigu, Tainan). M. lamarckii JML1 was commercially purchased from GOURMET HUNTER CO., LTD., a Taiwan-based international trading company specializing in aquatic products, originating from an aquaculture farm in Chiba, Japan. Genomic DNA was extracted from 25 mg of muscle tissue using the Nanobind® PanDNA Kit (PacBio, USA) following the ‘Extracting DNA from animal tissue using the Nanobind® PanDNA kit’ protocol. The extracted DNA was stored at −80 °C to preserve its integrity. DNA quality was assessed using 1.0% agarose gel electrophoresis, fluorescence quantification with the Qubit™ 4 Fluorometer (Thermo Fisher Scientific, USA) with Qubit™ dsDNA BR Assay Kits (Thermo Fisher Scientific, USA), as well as spectrophotometric analysis using the NanoDrop™ One Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific, USA).
Phylogenetic analysis of Meretrix species
There are 33 Cytochrome c oxidase subunit I (COXI) sequences from Meretrix species were selected for phylogenetic analysis, 27 sequences from NCBI database (M. lamarckii, M. lusoria, M. lyrate, M. meretrix, and M. petechialis) and six from this study (M. lamarckii JML1, JML2 and Meretrix sp. MF1, MT1, MT2, MT3). A neighbor-joining tree was constructed using MEGA version 11.0.138, with 1000 bootstrap replicates and the Tamura-Nei model.
Library preparation and sequencing
Genomic DNA was purified using AMPure XP Reagent (Beckman Coulter, USA) following the manufacturer’s protocol, and each purified sample was quantified using the Qubit™ 4 Fluorometer with Qubit™ dsDNA BR Assay Kits. Nanopore sequencing libraries were prepared using SQK-LSK110 Ligation Sequencing Kit (Oxford Nanopore Technologies, UK) according to the manufacturer’s protocol. A 150 µL aliquot of the library was loaded onto FLO-PRO002 (R9.4.1) flow cells (Oxford Nanopore Technologies, UK) for the PromethION 2 Solo (Oxford Nanopore Technologies, UK), and sequenced for approximately 120 hrs. The reads were then basecalled using Dorado version 0.7.0 (https://github.com/nanoporetech/dorado) with the super-accurate (SUP) model, yielding 80.02 Gb of data with 6.76 M high-quality reads for Meretrix sp. MF1 (Table 1). Additionally, the data for Meretrix sp. MT1 and M. lamarckii JML1 are summarized in Table 1. Illumina sequencing libraries were constructed using the TruSeq® Nano DNA Library Prep Kit (Illumina, USA) following the manufacturer’s guidelines. Genomic DNA was fragmented to approximately 350 bp via sonication, purified with Sample Purification Beads (Illumina, USA), and sequenced on the NovaSeq X Plus System (Illumina, USA), producing 150 bp paired-end reads. The raw Illumina reads, averaging 50.1 Gb per sample, were processed using fastp version 0.23.49 for quality control (Table 1). For chromosome-level assembly, the Hi-C library was constructed using the Dovetail® Omni-C® Kit (Cantata Bio, USA) following the manufacturer’s protocol. The library quality was assessed using a Qsep 100 Bio-Fragment Analyzer (BiOptic, Taiwan) with an S2 Standard Cartridge Kit (BiOptic, Taiwan) and a Qubit™ 4 Fluorometer with Qubit™ dsDNA HS Assay Kits (Thermo Fisher Scientific, USA). The library was then sequenced on the Novaseq X Plus System, generating 150 bp paired-end reads and yielding 46.48 Gb of data, with 309.86 M reads (Table 1).
Genome assembly and scaffolding
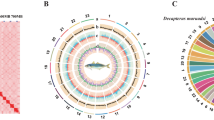
The general workflow of this study is illustrated in Fig. 1. Draft genome for Meretrix sp. MF1 and Meretrix sp. MT1 were generated using Nanopore data processed with Nextdenovo version 2.5.210. However, due to the shorter read lengths in M. lamarckii JML1 Nanopore data, its genome was assembled using Masurca version 4.1.211. The data were then processed with NanoFilt version 2.8.012 with Q12 for quality control. Next, both Nanopore and Illumina data were integrated and polished with Nextpolish version 1.4.113 followed by Purge_Dups version 1.2.614 to remove redundant sequences. Hi-C data was utilized to construct the chromosome-level genome assembly for Meretrix sp. MF1. Initially, fastp version 0.23.49 was employed for quality control, and Chromap version 0.2.715 was used for alignment and preprocessing. Scaffolding was carried out using YaHS version 1.2.216 to generate chromosome-level scaffolds. Subsequently, Juicer tools version 2.20.0017 was applied to construct the Hi-C contact matrix and contact map. The resulting chromosome-level genome assembly for Meretrix sp. MF1 had a total length of 883.3 Mb, with a longest scaffold of 59.29 Mb, an N50 of 46.87 Mb, and an L90 of 17 (Table 2). The Hi-C map (Fig. 2A) revealed 19 chromosome-scale scaffolds, which collectively accounted for 99.54% of the total genome size. Chromosome sizes ranged from 28.62 Mb to 59.29 Mb, with an average length of 46.27 Mb (Table 3). The genome was further visualized using TBtools-II version 2.15618 (Fig. 2B). To refine and scaffold the genomes of Meretrix sp. MT1 and M. lamarckii JML1, RAGTAG version 2.1.019 was used, with M. petechialis (GCA_046203225.1) serving as the reference genome for Meretrix sp. MT1, and Meretrix sp. MF1 as the reference for M. lamarckii JML1. Redundant sequences were then filtered using Purge_Dups version 1.2.614, and Nextpolish version 1.4.113 was applied for a final round of genome refinement. The final assembly details for all three species are summarized in Table 3.

Schematic overview of the general workflow.

Characteristics of Meretrix sp. MF1 genome assembly. (A) Hi-C heatmap of chromosomal interactions in the Meretrix sp. MF1 genome. (B) A circos plot of the Meretrix sp. MF1 genome, with tracks from innermost to outermost as follows: (a) Numbers and sizes of Meretrix sp. MF1 chromosomes; (b) Scatter plot of N ratio; (c) Line plot of GC skew; (d) Heatmap of gene density; (e) Bar plot of GC ratio.
Mitochondrial genome assembly
The mitochondrial genome was assembled using Illumina data with MitoZ version 3.620, which was further employed for mitochondrial annotation. To ensure accuracy, the assembled mitochondrial genome was compared against the nuclear genome using BLAST + version 2.16.021, and the verified mitochondrial sequence was incorporated into the final genome assembly. Notably, Meretrix sp. MF1 and Meretrix lamarckii JML1 exhibited the closest match to the same species, Meretrix lamarckii, albeit from distinct sources. Specifically, Meretrix sp. MF1 showed the highest similarity to Sequence ID: NC_016174.1 (GenBank), while Meretrix lamarckii JML1 showed the highest similarity to Sequence ID: KP244451.1. Furthermore, mitochondrial data revealed an additional tRNA-Leu in Meretrix sp. MF1 compared to Meretrix lamarckii JML1, potentially indicating distinct species status. In addition, Meretrix sp. MT1 was found to be most closely related to Meretrix lusoria (Sequence ID: NC_014809.1). A summary of all assembled mitochondrial data is provided in Table 4.
Repetitive sequence identification
RepeatModeler version 2.0.522 and RepeatMasker version 4.1.523 were used to analyze the Meretrix genome assemblies, enabling the de novo identification of transposable elements (TEs) and the classification of repetitive and low-complexity sequences (Table 5). The total proportion of repetitive elements in Meretrix sp. MF1, Meretrix sp. MT1, and M. lamarckii JML1 genomes were 41.57%, 41.75%, and 40.35%, respectively, with unclassified repeats accounting for 32.30%, 32.40%, and 31.36%. In terms of TE composition, Retroelements (Class I) were identified, constituting 6.99%, 6.55% and 6.64% of the genomes, respectively. The DNA transposons (Class II) were 1.98%, 1.59% and 1.77%, respectively. The consistent repeat content and distribution patterns across the three Meretrix species suggest a conserved genome organization and repetitive element dynamics within the genus.
Gene prediction and functional annotation
Gene prediction was performed on a genome version that was soft-masked for repeats using RepeatMasker version 4.1.523. The prediction was carried out with BRAKER version 3.0.824, employing a protein evidence-based approach using Metazoa dataset from OrthoDB version 1225. Gene prediction for Meretrix sp. MF1 was performed using BRAKER, which initially predicted 45,263 genes and 49,050 transcripts. To address gene over-prediction, the selectSupportedSubsets.py script within the BRAKER package was utilized. This script classifies predicted genes into three confidence categories: fully supported by hints (highest confidence), partially supported by hints, and not supported by hints (lowest confidence, purely computational). Subsequently, the selectSupportedSubsets.py script was employed to filter transcripts based on hint support, resulting in a subset of 32,329 transcripts. Transposable elements (TEs) were then masked using TEsorter version 1.2.726, yielding a final set of 30,417 transcripts. Functional annotation was conducted using EggNOG-mapper version 2.1.1227 and InterProScan version 5.73–104.028,29, to identify protein homologs, which included six database resources: eggNOG, Gene Ontology (GO) terms, Kyoto Encyclopedia of Genes and Genomes (KEGG), InterPro, Protein ANalysis THrough Evolutionary Relationships (PANTHER), and Pfam. A total of 25,531 genes were successfully annotated with functional information from at least one of these databases. Comprehensive gene annotation statistics for the Meretrix genome are provided in Supplementary Table 1.
Genomic similarity comparison and evolutionary analysis
FastANI version 1.3430 was applied to calculate the ANI among the genomes of Meretrix sp. MF1, Meretrix sp. MT1, M. lamarckii JML1, and M. petechialis. The results revealed that the ANI between Meretrix sp. MF1 and M. lamarckii JML1 was 94.33% (other comparisons are provided in Supplementary Table 2), suggesting that Meretrix sp. MF1 might represent a potentially novel species in Taiwan. We propose the name M. formosana. To explore evolutionary relationships, BUSCO version 5.8.331 was used to extract conserved Metazoa homologous genes from 11 genomes of Veneridae, including Callista chione, Cyclina sinensis32, Mercenaria mercenaria33, M. lamarckii JML1, M. petechialis6, Meretrix sp. MF1, Meretrix sp. MT1, Mysia undata, Ruditapes philippinarum32, Saxidomus purpurata34, and Venus verrucosa (Supplementary Table 3). Multiple sequence alignment was performed using MUSCLE version 5.335, followed by trimming with trimAI version 1.5.036 to generate the supermatrix alignment file. A phylogenetic tree was constructed based on the concatenated alignments using IQ-TREE version 1.6.1237, incorporating divergence times estimates obtained from the TimeTree database38 (accessed on Feb. 10, 2025). The estimated divergence times included 194 million years between M. mercenaria and V. verrucosa, 171 million years between V. verrucosa and R. philippinarum. The final phylogenetic tree was visualized using MEGA version 11.0.138, with M. mercenaria as the outgroup (Fig. 3). Genome-wide collinearity analysis was performed among M. lamarckii JML1, Meretrix sp. MF1, Meretrix sp. MT1, and M. petechialis using MCscanX version 1.0.039, then visualized with ChiPlot website (https://www.chiplot.online) (Fig. 4).

Estimated divergence times among Meretrix species inferred from metazoan orthologous genes. Phylogenetic tree of 11 mollusk species, rooted with Mercenaria mercenaria as the outgroup. Bootstrap values are shown in red next to each node. Divergence time estimates from the TimeTree database are indicated by blue. Estimated divergence times between species pairs are listed next to each node. Mya: million years ago.

Whole genome synteny and collinearity among Meretrix species. This figure displays the genome-wide collinearity among M. lamarckii JML1, Meretrix sp. MF1, Meretrix sp. MT1, and M. petechialis. Each block represents a distinct chromosome, and lines of the same color connect and highlight regions of collinearity between species.
Data Records
All raw sequencing data have been deposited in the BioProject at NCBI under accession number PRJNA122774040.
The Illumina data were deposited in the Sequence Read Archive at NCBI under accession number SRR32575144, SRR32575146, and SRR3257514941.
The Nanopore data were deposited in the Sequence Read Archive at NCBI under accession number SRR32575145, SRR32575147, and SRR3257515041.
The Hi-C data were deposited in the Sequence Read Archive at NCBI under accession number SRR3257514841.
The assembled genome were deposited in the Genbank under the accession number GCA_04924435542, GCA_04924436543, and GCA_04924437544.
The mitochondrial genome assembly under the accession number PV38317045, PV38317146, and PV38317247.
Genome annotation files are available in Figshare48.
Technical Validation
Genome assembly and annotation completeness evaluation
To assess the completeness and accuracy of the assembled genomes, multiple quality assessment tools were utilized. First, BUSCO version 5.8.331 with the mullsuca_odb12 lineage database, was used to evaluate the genome completeness. In the Meretrix sp. MF1 genome, 4264 (96.4%) single-copy ortholog were fully identified, while Meretrix sp. MT1 and M. lamarckii JML1 contained a complete set of 4116 (93.1%) and 4095 (92.6%) single-copy orthologs, respectively. The completeness scores for all three species exceeded 92.6% based on mullsuca_odb12 database, demonstrating the high quality and completeness of the assembled genomes (Table 6). Subsequently, BUSCO was applied with the mollusca_odb12 lineage database to assess the completeness of the predicted proteins. Results indicated that 4017 (90.9%) single-copy orthologs were fully identified in the Meretrix sp. MF1 predicted protei. In comparison, Meretrix sp. MT1 and M. lamarckii JML1 exhibited a complete set of 3752 (84.9%) and 3266 (73.9%) single-copy orthologs, respectively (Supplementary Table 4).
Next, Merqury version 1.349 was used to evaluate genome completeness using a k-mer-based approach. K-mers derived from Nanopore data were analyzed to calculate the quality value (QV) score, resulting in 97.62% k-mer completeness and an assembly consensus QV of 49.74 in Meretrix sp. MF1 (Supplementary Table 5). The statistical results for Meretrix sp. MT1 and M. lamarckii JML1 are also presented in Supplementary Table 5. To further assess assembly accuracy, Illumina reads were aligned to the genome using BWA version 0.7.1850. Statistical analysis with SAMtools version 1.2151 showed that 99.72% of the Illumina reads successfully mapped to the genome, achieving a coverage of 98.25%, confirming the high accuracy of the assembly (Supplementary Table 6). The results for Meretrix sp. MT1 and M. lamarckii JML1 are also presented in Supplementary Table 5. Omni-C library quality control was performed following the official Cantata Bio standard protocol (https://omni-c.readthedocs.io/en/latest/). The results yielded 151,321,804 total read pairs, with 58.36% mapped read pairs and 86.83% non-duplicate valid read pairs (cis ≥ 1 kb + trans). More detailed statistical information is presented in Supplementary Table 7. Additionally, Juicebox version 1.11.0852 was employed to visualize the assembled scaffolds and detect potential misassemblies. Manual inspection revealed no characteristic patterns of read coverage indicative of misjoins, translocations, or inversions.
Code availability
Genome annotation:
(1) RepeatModeler: parameters: all parameters were set as default.
(2) RepeatMasker: parameters: -e rmblast -lib database_repeat-families.fa genome.fasta -xsmall -s -gff.
(3) Braker3: parameters: --genome=genome.fa --prot_seq = proteins.fa --gff3.
Genome assembly:
(1) NextDenovo: parameters: job_type = local task = all rewrite = yes deltmp = yes parallel_jobs = 20 input_type = raw read_type = ont input_fofn = input.fofn read_cutoff = 1k genome_size = 1 g sort_options = -m 50 g -t 30 minimap2_options_raw = -t 8 pa_correction = 5 correction_options = -p 30 minimap2_options_cns = -t 8 nextgraph_options = -a 1
(2) Masurca: parameters: PE = pe 500 50 Illumina.fq.gz NANOPORE = nanopore.fastq EXTEND_JUMP_READS = 0 GRAPH_KMER_SIZE = auto USE_LINKING_MATES = 0 USE_GRID = 0 GRID_ENGINE = SGE GRID_QUEUE = all.q GRID_BATCH_SIZE = 500000000 LHE_COVERAGE = 25 LIMIT_JUMP_COVERAGE = 300 CA_PARAMETERS = cgwErrorRate = 0.15 CLOSE_GAPS = 1 NUM_THREADS = 40 JF_SIZE = 200000000 SOAP_ASSEMBLY = 0 FLYE_ASSEMBLY = 0
(3) NextPolish: parameters: job_type = local task = best rewrite = 1212 deltmp = yes rerun = 3 parallel_jobs = 2 multithread_jobs = 10 genome_size = auto polish_options = -p sgs_options = -max_depth 100 -bwa lgs_options = -min_read_len 1k -max_depth 100 lgs_minimap2_options = -x map-ont.
(4) Purge_dups: This tool was run with default parameters, without modifying its configuration file. The process followed these steps:
minimap2 -t 80 -x map-ont genome.fasta reads.fastq | gzip -c - > pb_aln.paf.gz
pbcstat pb_aln.paf.gz
calcuts PB.stat > cutoffs 2> calcults.log
split_fa genome.fasta > genome.fasta.split
minimap2 -t 80 -xasm5 -DP genome.fasta.split | pigz -c > genome.fasta.split.self.paf.gz
purge_dups -2 -T cutoffs -c PB.base.cov genome.fasta.split.self.paf.gz > dups.bed 2> purge_dups.log
get_seqs dups.bed $asm
Orthologous genes analysis:
(1) BUSCO: parameters: -i genome.fa -r -o Busco_result–lineage_dataset metazoan_odb12/mollsuca_odb12 -m geno/proteins -f–offline -augustus.
(2) iqtree: parameters: iqtree -s SUPERMATRIX -m TEST -bb 1000 -alrt 1000.
References
Lutaenko, K. A. Biodiversity of bivalve mollusks in the western South China Sea: an overview. Biodiversity of the western part of the South China Sea/eds AV Adrianov, KA Lutaenko. Vladivostok: Dalnauka, 315–384 (2016).
Chen, H.-C. Recent innovations in cultivation of edible molluscs in Taiwan, with special reference to the small abalone Haliotis diversicolor and the hard clam Meretrix lusoria. Aquaculture 39, 11–27 (1984).
Liu, B., Dong, B., Tang, B., Zhang, T. & Xiang, J. Effect of stocking density on growth, settlement and survival of clam larvae, Meretrix meretrix. Aquaculture 258, 344–349 (2006).
Lu, T. H., Yang, Y. F., Chen, C. Y., Wang, W. M. & Liao, C. M. Quantifying the impact of temperature variation on birnavirus transmission dynamics in hard clams Meretrix lusoria. Journal of Fish Diseases 43, 57–68 (2020).
Chang, C. C., Huang, J. F., Schafferer, C., Lee, J. M. & Ho, L. M. Impacts of culture survival rate on culture cost and input factors: Case study of the hard clam (Meretrix meretrix) culture in Yunlin County, Taiwan. Journal of the World Aquaculture Society 51, 139–158 (2020).
Law, S. T. S. et al. Genomes of two indigenous clams Anomalocardia flexuosa (Linnaeus, 1767) and Meretrix petechialis (Lamarck, 1818). Scientific data 12, 409 (2025).
Chen, C.-C. Phylogenetic analysis of Meretrix spp. based on Cytochrome c oxidase subunit I (COXI) gene sequences. Figshare https://doi.org/10.6084/m9.figshare.28674617 (2025).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Molecular biology and evolution 38, 3022–3027 (2021).
Chen, S. Ultrafast one‐pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2, e107 (2023).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biology 25, 107 (2024).
Zimin, A. V. et al. The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677 (2013).
De Coster, W., D’hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nature communications 12, 6566 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Molecular plant 16, 1733–1742 (2023).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome biology 23, 258 (2022).
Meng, G., Li, Y., Yang, C. & Liu, S. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic acids research 47, e63–e63 (2019).
Camacho, C. et al. BLAST+: architecture and applications. BMC bioinformatics 10, 1–9 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0 http://www.repeatmasker.org.RMDownload.html (2013).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Research 34, 769–777 (2024).
Tegenfeldt, F. et al. OrthoDB and BUSCO update: annotation of orthologs with wider sampling of genomes. Nucleic Acids Research 53, D516–D522 (2025).
Zhang, R.-G. et al. TEsorter: an accurate and fast method to classify LTR-retrotransposons in plant genomes. Horticulture Research 9, uhac017 (2022).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular biology and evolution 38, 5825–5829 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic acids research 49, D344–D354 (2021).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature communications 9, 5114 (2018).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Current Protocols 1, e323 (2021).
Xu, R. et al. Multi-tissue RNA-Seq analysis and long-read-based genome assembly reveal complex sex-specific gene regulation and molecular evolution in the Manila clam. Genome Biology and Evolution 14, evac171 (2022).
Farhat, S. et al. Comparative analysis of the Mercenaria mercenaria genome provides insights into the diversity of transposable elements and immune molecules in bivalve mollusks. BMC genomics 23, 192 (2022).
Kim, J. et al. Chromosome-level genome assembly of the butter clam Saxidomus purpuratus. Genome Biology and Evolution 14, evac106 (2022).
Edgar, R. C. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nature Communications 13, 6968 (2022).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular biology and evolution 32, 268–274 (2015).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40, e49–e49 (2012).
NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1227740 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP568055 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_049244355.1 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_049244365.1 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_049244375.1 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:PV383170.1 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:PV383171.1 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:PV383172.1 (2025).
Chen, C.-C. Annotation files for Meretrix genome assembly. Figshare https://doi.org/10.6084/m9.figshare.29145311 (2025).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome biology 21, 1–27 (2020).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Acknowledgements
This study was supported by the National Science and Technology Council of Taiwan (MOST 111-2628-M-019-001-MY3, and 113-2119-M-001-011-).
Author information
Authors and Affiliations
Contributions
Y.N.H. conceived and supervised the study. C.C.C., H.Y.L., T.H.H. and Y.N.H. collected the sample. C.C.C. performed the laboratory work. C.C.C. and Y.N.H. performed bioinformatics analysis. C.C.C. and H.Y.L. drafted the manuscript. T.H.H., S.L.T. and Y.N.H. provided review and modification of the manuscript. All authors read and approved of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, CC., Hsu, TH., Lu, HY. et al. High-quality chromosome-level genome of three Meretrix species using Nanopore and Hi-C technologies. Sci Data 12, 1141 (2025). https://doi.org/10.1038/s41597-025-05454-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05454-2
