
Abstract
Trabala vishnou (Lefèbvre, 1827) (Lepidoptera: Lasiocampidae) is a destructive leaf-eating pest that causes severe damage to forest ecosystems, leading to substantial economic losses. Herein, we sequenced and assembled a high-quality chromosome-level genome of T. vishnou using a combination of Illumina reads, PacBio HiFi reads, and High throughput Chromosome Conformation Capture (Hi-C) technologies. The genome size is 561.86 Mb and spans 25 chromosomes, exhibiting a high level of contiguity (scaffold/contig N50 = 21.75 Mb/20.67 Mb). Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis a 99.5% completeness score for this genome assembly. Repeat elements constitute 62.66% of the genome. A total of 1,630 non-coding RNAs and 12,895 protein-coding genes have been identified within the genome. The first chromosome-level genome of T. vishnou serves as a valuable reference for elucidating the evolution of functional traits in Lasiocampidae family and will facilitate the development of strategies for controlling defoliating pests.
Similar content being viewed by others

Background & Summary
Lepidoptera is the second largest order in Insecta, comprising moths and butterflies, with over 180,000 described species worldwide. Lasiocampidae Harris, 1841 is a large family of Lepidoptera, encompassing approximately 2,000 known species across 225 genera primarily distributed in tropical and subtropical regions1,2. Adults of the Lasiocampidae family are commonly known as lappet moths due to the distinctive wavy-edged hindwings that extend beyond both sides of the forewings when at rest, superficially resembling dried leaves. These adults are characterized by their medium to large size, broad triangular or rounded forewings, and subrotund hindwings, which lack the typical wing-coupling structures found in other moth families. Sexual dimorphism is evident, with females generally exhibiting a larger body size compared to males3. The eggs of Lasiocampidae species display a diverse range of morphologies, color patterns, and oviposition strategies4. The eggs are laid on host plant singly, in small group, or in large masses. They are broadly oval, with the micropyle at one pole, sometimes patterned, and with the chorion extensively sculptured in most species5. Lasiocampid larvae are commonly called hairy caterpillars, which are usually brightly coloured with the clothing of setae. The larvae are polyphagous, feeding on a wide range of tree leaves, mainly including Pinaceae, Fagaceae, Salicaceae, and Rosaceae. During period of large-scale outbreaks, their feeding can result in extensive defoliation of trees, leading to significant economic losses and ecological impacts3.
The genus Trabala Walker, 1856 currently consists of 40 species, most of which are primarily distributed in the Oriental region6,7. The bluish yellow lappet moth, Trabala vishnou (Lefèbvre, 1827), is the most widely distributed species within the genus Trabala, primarily occurring across South Asia, Southeast Asia, and southern China8. The larvae of T. vishnou constitute a serious leaf-eating pest (Fig. 1) that feed on a wide range of hosts, including sea buckthorn, oak, walnut, willow, apricot, apple, and crabapple9. The larvae typically consume the leaves of host plants, compromising tree growth and eventually causing tree mortality, thereby significantly impacting their ecological values and economic benefits. Although some studies have focused on the ecological, biological and morphological aspects of chemical and biological control9,10,11,12, scarce information is available regarding the valuable genomic resources of T. vishnou, hindering our understanding of the population genetics, phylogenetic relationships, and evolutionary history of Trabala species. Therefore, assembling a high-quality genome of T. vishnou could serve as a source of information to understand the origin and evolution of Lasiocampidae.

The life cycle of Trabala vishnou. (a) Eggs. (b) Larva. (c) Pupa. (d) Adult.
Here, we generated and assembled the first high-quality reference genome of T. vishnou by integrating Illumina sequencing data, PacBio HiFi reads, and High throughput Chromosome Conformation Capture (Hi-C) reads. The final genome size is 561.86 Mb with a contig N50 length of 20.67 Mb, assembling into 25 chromosomes. A total of 12,895 protein-coding genes were annotated in the chromosome-level genome assembly. This genome provides a valuable genomic resource for elucidating the evolutionary history of the genus Trabala and developing effective control strategies for these leaf-eating pests.
Methods
Sample collection and genome sequencing
Adult specimens of T. vishnou (Fig. 1) were collected from Mount Tianchi (34.26°N, 111.84°E; 1000 m), Songxian County, Luoyang Caity, Henan Province, China, in July 2024. The adults were reared in mesh cages under temperatures of 26 ± 1 °C and relative humidity of 30%–45%. Fertilized eggs were transferred into plastic containers for larval hatch. The female pupae were submerged in liquid nitrogen and then stored at −80 °C freezer for DNA extraction. All voucher specimens are deposited in the Biodiversity Museum, Henan University, Kaifeng, China (HENU).
Genomic DNA was extracted from a single female pupa utilizing the CTAB method. For short-read sequencing, a whole-genome library was constructed with the Agencourt AMPure XP-Medium kit and sequenced on the DNBSEQ-T7 platform. A PacBio HiFi library with an insert size of 20 Kb was prepared using the SMRTbell® Express Template Prep Kit 3.0(Pacific Biosciences #PN 101-853-100, CA, USA), followed by sequencing on the PacBio Sequel Revio platform. The Hi-C library was crafted through formaldehyde cross-linking, MboI restriction enzyme digestion, end repair of DNA fragments, DNA cyclization, and DNA purification, with final sequencing performed in 150 bp paired-end mode on the Illumina XPlus platform. Total RNA for second-generation transcriptome sequencing was extracted using the TRIzol™ Reagent kit. Library preparation was performed with the VAHTS mRNA-seq V2 Library Prep Kit and sequenced on the Illumina XPlus platform with 150-bp paired-end (PE) reads. The Oxford Nanopore transcriptome (RNA-ONT) sequencing was executed using the RNAprep Pure Plant Plus Kit (DP441) for RNA extraction. The library was constructed with the SQK-PCS109 and SQK-PBK004 kits and sequenced on the Oxford Nanopore PromethION platform.
Finally, a total of 41.60 GB of whole-genome sequencing (WGS) data, 41.42 GB of HiFi data, 50.79 GB of Hi-C data, 9.21 GB of RNA-seq data, and 11.42 GB of RNA-ONT data were generated, producing a combined dataset of 154.44 GB (Table 1).
Genome survey and assembly
The primary objective of genome survey analysis is to predict genome size, heterozygosity, and repeat sequence proportion, thereby facilitating the selection of appropriate assembly tools and adjustment of corresponding parameters for subsequent analyses. The BGI sequencing data were first processed using fastp v0.23.413 with parameters (‘-q 20 -D -g -x -u 10 -5 -r -c’) for quality control and trimming. Genome survey analysis was performed based on k-mer frequency distribution, where k-mer frequencies were assessed using khist.sh (a component of BBTools v38.9014) along with genome size estimation, with the sequence length set to 21 k-mer. The results revealed an estimated genome size of 564.01 Mb, with a heterozygosity rate of 1.81% and a repetitive sequence content of 58.33%. The proportion of repetitive sequences in the genome is relatively high, falling within the medium to high range of repetition.
High-quality HiFi reads were generated using pbccs v6.4.0 (https://github.com/PacificBiosciences/ccs). The initial assembly was conducted with Hifiasm v0.24.015 using default parameters. Owing to the exceptional base quality of the assembly, achieving a QV value exceeding 50, the conventional error-correction step (polishing) was deemed unnecessary. Furthermore, comprehensive evaluations using BUSCO v5.7.116 analyses demonstrated the high completeness of the genome assembly and the absence of significant heterozygosity, thereby eliminating the need for additional heterozygosity removal procedures.
The chromosome-level scaffolding was achieved using Hi-C data processed through the YAHS v1.217 pipeline. First, the Hi-C data underwent quality control with chromap v0.2.618, ensuring high-quality input for subsequent steps. Subsequently, two iterative rounds of scaffolding were performed using YAHS v1.2 with default parameters to generate the preliminary assembly. The initial scaffolding was meticulously corrected using Juicebox v1.11.0819 for structural anomalies, after which a final round of assembly was executed. The sequencing depth of each chromosome was meticulously evaluated using SAMtools v1.1020, with the input alignment BAM files generated by minimap2 v2.2421 using either HiFi reads (‘-ax map-hifi’) or second-generation WGS reads (‘-ax sr’).
The integrity of the genome was evaluated using BUSCO v5.7.1 with the insecta_odb10 reference dataset, which comprises 1,367 single-copy orthologous genes. The assembly was validated by mapping raw sequencing data from both the second-generation genome and transcriptome (short- and long-read data) back to the assembled genome. The mapping was conducted using minimap2 v2.24, and alignment rates were calculated using SAMtools for comprehensive quality assessment. Potential contamination within the assembly was identified using MMseqs. 2 v1322, performing a BLASTN-like search against the NCBI nt and UniVec databases. Additionally, the single-base quality score (QV value) was assessed with Merqury v1.323 to ensure high accuracy and reliability of the assembly.
Sex chromosomes and autosomes were distinguished by differences in sequencing depth, with the female sex chromosome exhibiting half the depth of autosomes. One chromosome showed significantly reduced sequencing depth compared to that of autosomes (Table 2). This chromosome was designated as the Z chromosome based on Hi-C interaction patterns. The W chromosome may have remained unassembled due to its highly repetitive sequence composition.
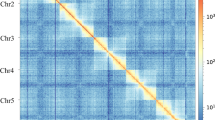
The final genome assembly spanned 561.86 Mb (114 scaffolds, 116 contigs), closely matching the predicted genome size (Table 3). Both the longest scaffold and contig extended to 35.23 Mb, with impressive scaffold and contig N50 values of 21.75 Mb and 20.67 Mb, respectively. The GC content was calculated at 36.39%, demonstrating the exceptional continuity and high quality of the assembly. The assembly successfully anchored 25 pseudochromosomes (Fig. 2), covering a total of 534.70 Mb of the genome, achieving an impressive anchoring rate of 95.16%. Notably, the base QV values for each chromosome exceeded 50 (Table 2), reflecting exceptional precision in the assembly quality. The BUSCO evaluation of the genome assembly revealed an exceptional completeness score of 99.5%, with only 0.7% of BUSCOs identified as duplicated. Alignment rates for BGI, HiFi, RNA-seq, and RNA-ONT data were remarkably high, reaching 98.74%, 100%, 93.11%, and 99.47%, respectively, all surpassing the 90% threshold (Table 3). The assembly achieved high standards in both continuity and completeness.

Hi-C interaction heatmap of the Trabala vishnou genome.
Repeat annotation
The software RepeatModeler v2.0.524 was employed with the enhanced LTR search option (‘-LTRStruct’) to construct a Trabala vishnou -specific de novo repeat library with the structural features of repetitive elements and de novo prediction methods. The artisanally crafted library was subsequently amalgamated with the Dfam 3.725 and RepBase-2018102626 databases to form the culminating comprehensive reference compendium for repetitive sequences. Repeat sequence identification was undertaken using RepeatMasker v4.1.527 in conjunction with the finalized repeat database. The analysis identified 2,148,681 repetitive elements totaling 352,087,259 bp (62.66% of the genome), in close agreement with computational predictions. Among these, the six most abundant repeat categories and their respective proportions were: LINEs (20.26%), SINEs (13.23%), Unknown (10.48%), LTRs (9.18%), DNA transposons (4.63%), and Rolling-circles (3.74%). To further explore the expansion origins of transposable elements (TEs), we utilized the script (‘calcDivergenceFromAlign.pl’) from RepeatMasker v4.1.5 to scrutinize the TE landscape. The results revealed that the transposable elements in the genome were primarily propelled by recent expansion events, emphasizing their dynamic evolutionary history (Fig. 3).

Transposable element landscapes of Trabala vishnou. (a) Interspersed repeat landscape based on K2P divergence. (b) Interspersed repeat landscape based on K2P divergence with unclassified repeats excluded.
Noncoding RNA annotation
The annotation of ncRNAs was performed using two complementary approaches: Initially, we used Infernal v1.1.528 to annotate rRNAs, snRNAs, and miRNAs by aligning sequences against the comprehensive non-coding RNA database (Rfam). Subsequently, tRNA sequences within the genome were predicted using tRNAscan-SE v2.0.1229, with low-confidence predictions filtered through the built-in script (‘EukHighConfidenceFilter’) to ensure the robustness and reliability of the results.
In summary, the genome annotation revealed a diverse array of 1,630 non-coding RNA elements, including rRNAs (312), miRNAs (589), snRNAs (179), tRNAs (399), ribozymes (3), and lncRNAs (2). Notably, the snRNAs encompassed spliceosomal RNAs (116), minor spliceosomal RNAs (13), C/D box snoRNAs (46), and HACA-box snoRNAs (4).
Protein-coding gene prediction and annotation
The comprehensive prediction of protein-coding gene structures was performed using the MAKER v3.01.0430 pipeline, with a sophisticated analytical workflow constructed by integrating three complementary types of evidence. First, during the initial data processing phase, we used HISAT2 v2.2.131 to align RNA-seq short-read data to the genome, generating BAM alignment files as transcriptomic evidence. Additionally, protein sequences of arthropods were extracted from the OrthoDB11 database32, and genomic data, protein sequences, and annotation files from five related species (Drosophila melanogaster, Manduca sexta, Amyelois transitella, Bombyx mori, and Spodoptera frugiperda) were collected to function as homology-based evidence. During the gene structure prediction phase, a multifaceted concurrent analysis was employed. The BRAKER v3.0.633 was used to integrate transcriptomic BAM files and protein sequences from the OrthoDB11 database, automatically training prediction models in Augustus v3.4.034 and GeneMark-ETP35. Simultaneously, GeMoMa v1.936 was employed to predict gene structures based on homology with closely related species and conservation of intron positions. The parameters were configured as GeMoMa.c = 0.4 and GeMoMa.m = 85000 to optimize the analysis. The results from these two ab initio prediction approaches were merged to generate a file, which functioned as input for the MAKER pipeline. Meanwhile, StringTie v2.2.137 was used to perform reference-guided assembly of the transcriptomic BAM files, generating gene structure predictions at the transcript level. The MAKER pipeline predicted a total of 12,895 protein-coding genes, with an average gene length of 20,940.5 bp. On average, each gene contained 7.7 exons with an average exon length of 314.6 bp, and 8.0 introns with an average intron length of 2,789.69 bp. Additionally, each gene encoded 7.6 coding sequences (CDS), with an average CDS length of 221.3 bp (Table 4). The predicted protein-coding gene sequences were assessed for completeness using BUSCO, revealing C:99.4% [S:85.9%, D:13.5%], F:0.0%, M:0.6%, and n:1367 consistent with the genome-level completeness assessment of 99.5%.
Gene functional annotations were performed utilizing Diamond v2.1.7.16138 in its highly sensitive mode (‘–very-sensitive -e 1e-5’) to query the UniProtKB database. Simultaneously, InterPro 5.70-102.039 was employed to search the Pfam database40, while eggNOG-mapper v2.1.1241 was utilized to explore the eggNOG v5.0.2 database42. Circos plots integrating genomic structure and annotations were generated using TBTools-II v2.09643 (Fig. 4). The predictions derived from these two approaches were subsequently integrated to generate the final comprehensive gene functional annotations. A total of 13,889 genes (97.71%) corresponded to records in the UniProtKB database. InterPro identified protein domains for 11,694 protein-coding genes, while InterPro and eggNOG-mapper together annotated 9,530 genes with Gene Ontology (GO) terms and 4,649 genes with KEGG pathway entries.

Circos plot of the Trabala vishnou genome. From the outer ring to the inner ring, the tracks represent chromosome length, GC content, gene density, DNA transposon density, SINE transposon density, LINE transposon density, and LTR transposon density, respectively.
Data Records
The raw sequencing data and the assembled genome of Trabala vishnou had been deposited in the NCBI database under the BioProject accession number PRJNA1229088. The corresponding RNA-ONT, Hi-C, RNA-seq, HiFi, and whole-genome sequencing (WGS) data can be accessed through the Sequence Read Archive (SRA) under the accession numbers SRR3287743844, SRR3287743945, SRR3287744046, SRR3287744147, and SRR3287744248, respectively. Additionally, the assembled genome had been submitted to GenBank and is available under the accession number GCA_050084765.149. The assembled genome and annotation data have been deposited in the Figshare database50 to facilitate open access and further research utilization.
Technical Validation
We performed a comprehensive quality assessment of the assembled genome using two complementary approaches. Initially, we evaluated the assembled genome using BUSCO v5.7.1 based on the insecta_odb10 reference dataset, including 1,367 single-copy ortholog genes. The final genome assembly displayed a BUSCO completeness of 99.5% (98.8% single-copy and 0.7% duplicated), 0.1% fragmented, and 0.4% missing BUSCOs. Subsequently, we aligned BGI, HiFi, RNA-seq short-read, and RNA-ONT data to the genome assembly to assess the utilization of raw data and the completeness of the assembly. The results showed alignment rates of 98.74%, 100%, 93.11%, and 99.47%, respectively. These assessments confirm the high quality of the genome assembly, demonstrating exceptional continuity and completeness. We employed BUSCO v5.7.1 to assess the completeness and redundancy of the predicted protein-coding gene sequences. The assessment yielded C:99.4% [S:85.9%, D:13.5%], F:0.0%, M:0.6%, closely matching the genome-level completeness estimate of 99.5%.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding software.
References
Van Nieukerken, E. J. et al. Order Lepidoptera Linnaeus, 1758. In: Zhang, Z.-Q. (Ed.) Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. Zootaxa. 3148, 212–221 (2011).
Prozorov, A. M. et al. A new genus of Afrotropical Lasiocampini: Revaya gen. n. (Lepidoptera, Lasiocampidae, Lasiocampinae). Zootaxa. 5369, 207–222 (2023).
Liu, Y. Q. & Wu, C. S. Fauna Sinica. Insecta. Volume 47: Lepidoptera. Lasiocampidae. Beijing, China: Science Press, pp 1–385 (2006).
Farrar, N., Zamani, A. A., Naghadeh, N. M., Haghani, M. & Azizkhani, E. Dynamism, biology and morphology of jujube lappet moth Streblote siva (Lepidoptera: Lasiocampidae) in Bushehr city. Plant Prot. Sci. J. Agric. 41, 11–26 (2018).
Zolotuhin, V. V. & Kurshakov, P. A. Fine sculpture and phylogenetic implications of egg shell morphology in the Lasiocampidae (Lepidoptera, Lasiocampidae). Neue Entomol. Nachr. 67, 3–21 (2011).
GBIF Secretariat. GBIF Backbone Taxonomy. Checklist dataset https://doi.org/10.15468/39omei (2023).
Saini, S., Kaleka, A. S. & Singh, D. Studies on three species of genus Trabala Walker (Lasiocampidae: Bombycoidea: Lepidoptera) from north west India. Ann. Entomol. 37, 19–27 (2019).
Liu, Y. H. et al. Effects of short-term high temperatures on survival and reproduction of Trabala vishnou gigantina Yang (Lepidoptera: Lasiocampidae). Pakistan J. Zool. 54 (2021).
Liu, Y. H. et al. Damage of Trabala vishnou gigantina (Lepidoptera: Lasiocampidae) to the sea-buckthorn forest and its biological characteristic. Plant Prot. 39, 147–151 (2013).
Kakulte, V. R. & Mamlayya, A. B. Insect pests of black plum, Syzygium cumini (l) skeels from amba reserve forest of Kolhapur district, Maharashtra. J. Entomol. 7, 31–36 (2022).
Huang, S. X., Li, J. L., Qin, X. J., Wang, Z. W. & Hou, B. Toxicity of four biopharmaceutics for controlling Trabala viahnou gigantina Yang larvae in blueberry. Journal of Anhui Agri. Sci. 43, 144–145, 147 (2015).
Chen, Y. X. et al. Bioactivity of 14 insecticides against the 4th instar larvae of Trabala vishnou gigantina. J. Northwest For. Univ. 37, 170–175 (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Bushnell, B. BBMap., Available online: https://sourceforge.net/projects/bbmap/.
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and streamlined workfows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: Yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 12, 6566 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Steinegger, M. & Söding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457 (2020).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0., Available online: http://www.repeatmasker.org (2013/2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Kuznetsov, D. et al. OrthoDB V11: Annotation of orthologs in the widest sampling of organismal diversity. Nucleic Acids Res. 51, D445–D451 (2023).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, lqaa108 (2021).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP: GeneMark-ETP signifcantly improves the accuracy of automatic annotation of large eukaryotic genomes. Genome Res. 34, 757–768 (2024).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19, 189 (2018).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Paysan-Lafosse, T. et al. InterPro in 2022. Nucleic Acids Res. 51, D418–D427 (2023).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Chen, C. et al. TBtools-II: A ‘one for all, all for one’ bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32877438 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32877439 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32877440 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32877441 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32877442 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_050084765.1 (2025).
Pu, Y. L. Chromosome-level genome assembly and annotations of Trabala vishnou (Lepidoptera, Lasiocampidae). Figshare. https://doi.org/10.6084/m9.figshare.28638698 (2025).
Acknowledgements
We are grateful to Jia-Xin Xu, Jie Yang, Jia-Shuo He, He Zhu, and Xin-Rui Zhao for assistance in collecting specimens. We thank Prof. Feng Zhang for his constructive comments on the process of genome annotation. Special thanks to Yu Shi for his professional English editing of this manuscript. This research was supported by the National Natural Science Foundation of China (No. 32300379).
Author information
Authors and Affiliations
Contributions
K.G. conceived of the study. Y.L.P. conducted the data analysis. Y.L.P., T.Z.Y., and H.R.W. wrote the manuscript. K.G. and H.P.Z. revised the manuscript. All authors participated in the development of this manuscript and provided their final approval.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pu, YL., Yan, TZ., Wang, HR. et al. The first chromosome-level genome of the lappet moth Trabala vishnou (Lepidoptera: Lasiocampidae). Sci Data 12, 1154 (2025). https://doi.org/10.1038/s41597-025-05456-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05456-0
