
Abstract
Batocera rufomaculata (Cerambycidae: Lamiinae), a prominent representative of longhorned beetles, is a globally significant stem-boring pest, infesting over 50 species of deciduous trees. Despite its substantial ecological and economic impact, the genomic basis underlying its host adaptation remain poorly understood. Here, we present a chromosome-level genome assembly of B. rufomaculata, constructed using a combination of Illumina, PacBio HiFi, and Hi-C sequencing data. The genome spans 338.08 Mb, with a scaffold N50 of 37.00 Mb, and is organized into 10 pseudo-chromosomes, including a chromosome X validated by genome collinearity and sequencing depth analyses. Repetitive elements constitute 27.89% of the genome, totaling 94.29 Mb. Out of 17,887 predicted genes, 12,729 were functionally annotated with at least one supporting evidence. The high-quality genome assembly and annotation were confirmed by multiple metrics, including genome size, reads mapping rate (>99.5%), BUSCO completeness (>97.1%), and collinearity analyses. This comprehensive genomic resource provides a foundation for investigating the ecological adaptation of B. rufomaculata and offers valuable insights into the genetic mechanisms that could inform pest management strategies.
Similar content being viewed by others


Background & Summary
Longhorned beetles (Cerambycidae) are among the most ecologically significant and morphologically charismatic insects. They are distinguished by their prominent long antennae (1–2 times the length of their body) and are one of the most species-rich families, with approximately 35,000 extant species globally distributed1,2,3. These beetles exhibit remarkable ecological versatility, serving roles as decomposers, pollinators, vectors, and pests, while also displaying a wide phytophagous host range1,2,3. Batocera rufomaculata (Cerambycidae: Lamiinae) is a quintessential representative of this family. Known commonly as the mango stem borer, fig borer, or tropical fig borer due to its destructive tendencies4, this species poses a significant threat as a stem-boring pest in both non-wood product and timber forests. It infests over 50 deciduous tree species, including economically important crops such as Mangifera indica (mango), durian, Coffee, Morus spp., Moringa spp.5,6. Its distribution spans across Asia and Europe, with reported occurrences in countries such as India, Nepal, Pakistan, Thailand, Malaysia, China, Israel, Turkey, and France5,7,8,9. Despite its ecological and economic importance, the genomic basis underlying its host adaptation remain poorly understood.
Throughout its life cycle, B. rufomaculata poses a persistent threat to its host plants (Fig. 1), yet effective management strategies remain limited. A single female can lay up to 200 eggs onto the bark10. Once the larvae hatch, they burrow deep into the stems or shoots, making them difficult to control10. The larval stage lasts approximately one year, after which the insects pupate and emerge as adults with a lifespan of 60–100 days. Adults feed on green growth tips and twig bark, further damaging the host plants10,11. Current control methods primarily rely on physical or mechanical removal of the pests, which is both costly and inefficient10. However, advancements in molecular genetics and genomics offer promising avenues for pest management12. Techniques such as RNA interference (RNAi) and CRISPR/Cas9 have demonstrated significant potential in controlling invasive insect species13,14. Additionally, the decreasing costs of genomic sequencing technologies provide opportunities to unravel the molecular basis of pest evolution and outbreaks. Despite these advancements, no comprehensive genome resource is currently available for B. rufomaculata, hindering research into its biology and the development of targeted control measures.

Overview of Batocera rufomaculata (Degeer, 1775). Photos and permission by Yongke Zhang and Tianqi Bai. (a) Frass holes and larval excrement around the infected trunk of Mangifera indica. (b) Emergence holes created by adult on the trunk of Mangifera indica. (c) Longitudinal section of larval galleries within the trunk of Mangifera indica. (d) Egg. (e) Pupa. (f) Adult.
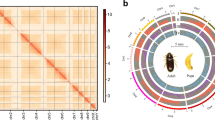
In this study, we present a high-quality chromosome-level genome assembly of B. rufomaculata, integrating PacBio high-fidelity (HiFi) reads, Illumina short-reads, and high-throughput chromosome conformation capture (Hi-C) data. The genome spans 10 pseudo-chromosomes (Fig. 2a,b), with chr10 identified as the X chromosome based on collinearity analysis and sequencing depth (Fig. 2c,d). The assembly and annotation were rigorously validated through metrics such as genome size comparison, read mapping rates, Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis, and synteny assessment (Tables 1–7). This high-confidence genomic resource provides a foundation for understanding the adaptive and demographic processes of B. rufomaculata and paves the way for the development of novel, targeted pest management strategies.

Assembly of the chromosome-level genome of Batocera rufomaculata. (a) The heatmap displays all interactions among 10 chromosomes. The color of the Hi-C interaction linkages represents the frequency, which ranges from yellow (low) to red (high). (b) Blocks on the outmost circle represent all 10 chromosomes. Peak plots from inner to outer circles represent gene density, repeat density, GC ratio, and the coverage of Hi-C reads, HiFi-reads, and Illumina short-reads of each chromosome, respectively (sliding window size = 50 Kb). (c) Mean sequencing depth of Illumina short-reads and PacBio HiFi-reads for each chromosome of B. rufomaculata genome. (d) Chromosome-level synteny analysis of the B. rufomaculata (Bru) and another two Cerambycidae beetles, Leptura quadrifasciata (Lqu) and Rutpela maculata (Rma).
Methods
Sample information
Samples of B. rufomaculata were collected in May 2023 from a mango plantation in Lujiang Township, Longyang District, Baoshan City, Yunnan Province (24°48’ N, 98°40’ E, 705 m above sea level) for this study. The collected samples were then loaded into 50 mL centrifuge tubes, quick-frozen with liquid nitrogen, and stored in an ultra-low-temperature refrigerator at −80 °C until use. Voucher specimens were deposited in the Tropical and Subtropical Cash Crop Research Institute, Yunnan Academy of Agricultural Sciences.
Illumina, PacBio HiFi, Hi-C, and RNA sequencing
For dissecting the reference genome of B. rufomaculata, genomic DNA was extracted from a male individual and prepared by the CTAB method followed by purification with a Grandomics Genomic kit for regular sequencing, according to the standard operating procedure provided by the manufacturer. DNA quality was monitored on 1% agarose gels, detected using NanoDrop™ One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA), and measured by Qubit® 4.0 Fluorometer (Invitrogen, USA). All sequencing was performed at NextOmics (Wuhan, China) (Table 1).
The short-reads libraries (350-bp insert size) were generated using Truseq Nano DNA HT Sample Preparation Kit (Illumina USA) following the manufacturer’s recommendations. The DNA fragments were then end-polished, A-tailed, and ligated with the full-length adapter for Illumina sequencing, followed by PCR amplification. 150-bp paired-end reads were generated on the Illumina NovaSeq 6000 platform. The quality per base was evaluated with FastQC15. Both low-quality reads ( > 90% bases with quality < Q30) and adaptor sequences were removed. A total of 89 Gb clean data was obtained.
The HiFi-reads libraries were prepared using SMRTbell® prep kit 3.0 kit following the manufacturer’s recommendations. After DNA quality control, DNA shearing and cleanup, DNA repair and A-tailing reactions, adapter ligation and cleanup, nuclease treatment, and size selection, the cleanup of libraries transferred to the PacBio Revio equipment according to the operating manual provided by PacBio. The min passes = 3 and min RQ = 0.99 default parameters in CCS software (https://github.com/PacificBiosciences/ccs) were utilized to generate 21 Gb high-precision HiFi reads with quality over Q20.
The Hi-C libraries for Illumina sequencing were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) according to the manufacturer’s instructions. During the process, the collected 106 cells were cross-linked, and the restriction enzyme DpnII was used to lyse and digest the isolated cells. The final library was sequenced on the Illumina Novaseq 6000 platform. After quality control, 61 Gb clean data were generated for Hi-C assembly.
Total RNA was also extracted from the same individual for extracting genomic DNA. The RNA libraries were generated using TruSeq RNA Library Preparation Kit (Illumina, USA) following the manufacturer’s recommendations. After cluster generation, the library preparations were sequenced on an Illumina Novaseq 6000 platform. About 6 Gb paired-end reads (150-bp) were generated and used to annotate the protein-coding genes of the genome of B. rufomaculata.
Chromosome-level genome assembly
The genome was assembled using hifiasm v0.20.0-r63916 with the parameter (‘-l 3 -n 4’). The haploids and contig overlaps were removed using purge_dups (https://github.com/dfguan/purge_dups). The non-redundant genome was polished using the HiFi reads and Illumina short-reads by NextPolish2 v0.2.117. Next, we pre-processed the Hi-C data with Juicer v1.618 and 3D-DNA v18011419. Manually visualized and corrected the pseudo-chromosomes with JuiceBox v2.15 (https://github.com/aidenlab/Juicebox). Finally, we ran 3D-DNA again to obtain the final chromosome-level genome assembly. The final genome size was 338.08 Mb, with a scaffold N50 of 37.03 Mb (Table 2), and 99.77% of the sequence anchored into 10 pseudo-chromosomes.
Repeat annotation
A combination of de novo and homology-based approaches was applied to predict the repeated sequences in the B. rufomaculata genome. RepeatModeler (http://www.repeatmasker.org/RepeatModeler/) was used to identify and classify novel TEs. The homology-based approach was used to detect known transposable elements (TEs) at DNA and protein levels. RepeatMasker was used to identify the DNA level TEs with the Repbase TE library 21.0420. RepeatProteinMask21 in RepeatMasker was used to search the protein level TEs with parameters “-noLowSimple -pvalue 0.0001”. In addition, Tandem Repeat Finder22 was used to discover the tandem repeats with parameters “2 7 7 80 10 50 2000 –d –h”. In total, we identified 94.29 Mb of repetitive elements, accounting for 27.89% of the genome (Table 3). Among these elements, 24.93% were identified as TEs, including the most abundant DNA transposons, followed by LTR, LINE, and SINE retrotransposons (Table 4).
Gene structure prediction
The protein-coding genes (PCGs) of the repeat-masked genome were predicted based on de novo prediction, homology prediction, and RNA-seq prediction. For de novo prediction, AUGUSTUS23 was used to predict coding genes. For homology prediction, protein data sets of other four long-horned beetles, Anoplophora glabripennis (GCF_000390285.2), Monochamus alternatus (GCA_035320865.1), Leptura quadrifasciata (GCA_963675865.1), and Rutpela maculata (GCA_936432065.2), were aligned against the assembled genome using TBLASTN with an E-value cut-off of 1e-5. Solar software (The Beijing Genomics Institute development) was used to conjoin the BLAST hits and GeneWise24 was applied to predict gene structures based on each BLAST hit. For RNA-seq prediction, we aligned RNA-seq using HISAT2 v2.2.125 and assembled unigenes using StringTie v2.1.626. TransDecoder (https://github.com/TransDecoder/TransDecoder) was used to predict the gene models. Through integration with EvidenceModeler v1.1.127, we predicted a total of 17,887 PCGs in the genome.
Gene function prediction
The function of predicted genes was annotated using BLASTP (E-value < 1e-05) against databases including SwissProt, TrEMBL, and NCBI non-redundant proteins database (NR). The structural domains and motifs of all genes were scanned against SMART, ProDom, Pfam, PRINTS, PROSITE, and PANTHER databases using InterProScan v5.2528. The Gene Ontology (GO) terms were extracted based on the corresponding InterPro entry. The metabolic pathways in which the genes might be involved were assigned by BLAST (E-value < 1e-05) against the Kyoto Encyclopedia of Genes and Genomes (KEGG) protein database29. In total, 12,729 predicted genes were supported by at least one functional clue (Table 5).
Data Records
All sequencing data (Short-reads, HiFi-reads, Hi-C reads, and RNA-seq) used for genome assembly and annotation, were submitted to NCBI under the BioProject PRJNA1209233. Illumina sequencing data for genome survey were deposited in the Sequence Read Archive at NCBI under accession number SRR3245864130. Hi-C sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRR3245640131. PacBio HiFi sequencing data were deposited in the Sequence Read Archive at NCBI under accession number SRR3245600932. RNA-seq data were deposited in the Sequence Read Archive at NCBI under accession numbers SRR3245863833. The final chromosome assembly was deposited in GenBank under accession number JBKOFD00000000034. The final gene annotation results of B. rufomaculata have been deposited in Figshare repository under a DOI number of https://doi.org/10.6084/m9.figshare.28466675.v135.
Technical Validation
Genome assembly and annotation assessment
The assembled size is slightly larger than the size of 325.90 Mb estimated by analyzing 17-mer frequency36 utilizing Illumina short-reads (Table 6). We segmented the assembled chromosomes into 50 kb-sized fragments and plotted a heatmap using juicerbox 2matrix (https://github.com/yukaiquan/biotools/tree/main/Hic/juicerbox2matrix). The heatmap shows that on all 10 chromosomes, the interaction strength at diagonal positions is significantly higher than at non-diagonal regions, indicating good quality of chromosome assembly (Fig. 2a). BUSCO v5.2.237 was used to evaluate the completeness and redundancy of gene regions of the assembly and the protein-coding genes based on the lineage dataset of endopterygota_odb10. The complete BUSCOs of draft genomes, final genomes, and PCGs have reached over 97.00% (Table 7).
The Illumina short-reads, HiFi-reads, and Hi-C reads were mapped onto the assembled genome using BWA v0.7.1238 with default settings. All mapping rates counted using the Samtools v1.2.139, have exceeded 99.50% (Table 1). The sequencing depths of chr10 had half the sequencing depth of the other chromosomes (Fig. 2c), suggesting chr10 may be the sex chromosome X.
Genome synteny
The chromosome X has been identified in the genome of L. quadrifasciata40 and R. maculata41 of the same family as B. rufomaculata. Collinearity analysis was performed by Python MCScanX pipeline42 in TBtools-II43. These three species showed good chromosomal collinearities and chr10 of B. rufomaculata has a good collinearity with chromosome X of L. quadrifasciata and R. maculata (Fig. 2d), confirming that chr10 should be sex chromosome X.
Code availability
All software and pipelines were executed following the manuals and protocols provided by the published bioinformatic tools. The version and parameters of the software have been described in the Methods section.
References
Lawrence, J. Coleoptera Synopsis and classification of living organisms. 2(SR) pp. 482–553 (1982).
Allison, J., Borden, J. & Seybold, S. A review of the chemical ecology of the Cerambycidae (Coleoptera). Chemoecology 14, 123–150, https://doi.org/10.1007/s00049-004-0277-1 (2004).
Rossa, R. & Goczał, J. Global diversity and distribution of longhorn beetles (Coleoptera: Cerambycidae). Eur Zool J 88(1), 289–302 (2021).
Mitra, B. et al. First record of Batocera rufomaculata (De Geer, 1775) from Sunderban Biosphere Reserve, West Bengal. Int J Ent Res. 1(3), 31–32 (2016).
Kariyanna, B. et al. Host plants record and distribution status of agriculturally important longhorn beetles (Coleoptera: Cerambycidae) from India. Progressive Research. 12(1), 1195–1199 (2017).
Sudhi-Aromna, S. et al. Studies on the biology and infestation of stem borer, Batocera rufomaculata, in durian. Acta Hortic. 787, 331–337, https://doi.org/10.17660/ActaHortic.2008.787.41 (2008).
Özdikmen, H. A complete list of invasive alien longhorned beetles species for Turkey (Coleoptera: Cerambycidae). Mun Ent Zool. 12(2) (2017).
Zhang, Y. et al. Batocera rufomaculata (Degeer), an important pest on Moringa spp. Forest Pest and Disease 38(03), 1–5, https://doi.org/10.19688/j.cnki.issn1671-0886.20180023 (2019).
Yang, H. F. et al. Scanning electron microscopic observations of the antennal sensilla of Batocera rufomaculata (Degeer) (Coleoptera: Cerambycidae). J Environ Entomol. 46(02), 530–539, https://doi.org/10.3969/j.issn.1674-0858.2024.02.25 (2024).
Magar, B. R., Joshi, M. & Poudel, S. Mango stem borer: a serious pest and management strategies. Reviews in Food and Agriculture. 3(2), 54–57, https://doi.org/10.26480/rfna.01.2022.24.27 (2022).
Manoj, K. et al. Morphomolecular study on the flat-faced longhorn beetle Batocera rufomaculata (De Geer, 1775) from Rehla, Palamu, Jharkhand, India. Notulae Scientia Biologicae. 16(4), 12079–12079, https://doi.org/10.15835/nsb16412079 (2024).
Kirk, H., Dorn, S. & Mazzi, D. Molecular genetics and genomics generate new insights into invertebrate pest invasions. Evol Appl. 6(5), 842–856, https://doi.org/10.1111/eva.12071 (2013).
Zuo, Y. et al. Genome mapping coupled with CRISPR gene editing reveals a P450 gene confers avermectin resistance in the beet armyworm. PLoS Genet. 17(7), e1009680, https://doi.org/10.1371/journal.pgen.1009680 (2021).
Buer, B. et al. Superior target genes and pathways for RNAi‐mediated pest control revealed by genome‐wide analysis in the beetle Tribolium castaneum. Pest Manag Sci https://doi.org/10.1002/ps.8505 (2024).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 27(6), 863–864, https://doi.org/10.1093/bioinformatics/btr026 (2011).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18(2), 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Hu, J. et al. NextPolish2: A repeat-aware polishing tool for genomes assembled using HiFi long reads. GPB. 22(1) https://doi.org/10.1093/gpbjnl/qzad009 (2024).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3(1), 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356(6333), 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Bao, W., Kojima, K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. Chapter 4, 4101–4114, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2), 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Hoff, K. J. & Stanke, M. WebAUGUSTUS–a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 41(Web Server issue), W123–W128, https://doi.org/10.1093/nar/gkt418 (2013).
Birney, E. & Durbin, R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 10(4), 547–548, https://doi.org/10.1101/gr.10.4.547 (2000).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 12(4), 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33(3), 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Haas, B. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9(1), R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 30(9), 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Kanehisa, M. and S. Goto, KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32458641 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32456401 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32456009 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32458638 (2025).
He, J., T. Bai, and X. Li. Chromosome-level genome assembly of Batocera rufomaculata (Degeer, 1775) using PacBio HiFi and Hi-C sequencing. Available from: https://identifiers.org/ncbi/insdc:JBKOFD000000000 (2025).
He, J. W. Chromosome-level genome assembly of Batocera rufomaculata (Degeer, 1775) using PacBio HiFi and Hi-C sequencing. figshare https://doi.org/10.6084/m9.figshare.28466675.v1 (2025).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv: Genomics, 1308.2012, https://doi.org/10.48550/arXiv.1308.2012 (2013).
Manni, M. et al. BUSCO Update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 38(10), 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25(14), 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16), 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Mitchell, R. et al. The genome sequence of the four-banded longhorn beetle, Leptura quadrifasciata Linnaeus, 1758. Wellcome Open Research. 9, 386, https://doi.org/10.12688/wellcomeopenres.22611.1 (2024).
Sivell, O. et al. The genome sequence of a longhorn beetle, Rutpela maculata (Poda, 1769). Wellcome Open Research. 8, 579, https://doi.org/10.12688/wellcomeopenres.20500.1 (2023).
Tang, H. et al. Synteny and collinearity in plant genomes. Science. 320(5875), 486–488, https://doi.org/10.1126/science.1153917 (2008).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol Plant. 16(11), 1733–1742, https://doi.org/10.1016/j.molp.2023.09.010 (2023).
Acknowledgements
We appreciate Yongke Zhang from the Yunnan Institute of Tropical Crops for providing a photo of the B. rufomaculata egg. This work was supported by grants from National Key R&D Program of China (Grant No. 2022YFC2602500), Yunnan Provincial Science and Technology Department (Talent Project of Yunnan: 202105AC160039;Yunnan Fundamental Research Projects: 202401BC070017; Yunnan Major Science and Technology Project: 202402AE090008), Sci-Tech Service Station of Farmer Academician in Huaping County (Document No. 9 of 2023 jointly issued by Yunnan Association for Sci-Tech), and Yunnan Academy of Agricultural Sciences Scientific Research Pre-Research Project (2024KYZX-07).
Author information
Authors and Affiliations
Contributions
L.X.Y. conceived and supervised the study. B.T.Q. and D.Z.W. collected samples. W.W.T. and W.Y.J. performed sequencing. H.J.W. performed genome assembly and annotation analysis. H.J.W. and B.T.Q. wrote the draft manuscript. H.J.W., L.X.Y, B.T.Q. and Z.H.R. improved and revised the manuscript. All authors improved, revised, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, J., Bai, T., Wan, W. et al. Chromosome-level genome assembly of stem borer Batocera rufomaculata using PacBio HiFi and Hi-C sequencing. Sci Data 12, 1099 (2025). https://doi.org/10.1038/s41597-025-05463-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05463-1
