
Abstract
In this study, we report a high-quality chromosome-level genome assembly of Actinidia chinensis var. chinensis ‘Guimi No. 2’. This cultivar, discovered in Guizhou karst ecosystems, exhibits resistance to Pseudomonas syringae pv. actinidiae (Psa). Using a combination of MGI short-read sequencing, PacBio HiFi long-read sequencing, and Hi-C technology, we generated a genome assembly of 608.43 Mb with a contig N50 of 20.70 Mb, and 99.70% of the assembly was successfully anchored onto 29 pseudochromosomes. The quality value (QV) and the LTR Assembly Index (LAI) of the assembled genome were 72.23 and 10.10. The BUSCO analysis indicated that the genome assembly and gene model prediction were 98.40% and 96.56% complete, respectively. A total of 251.15 Mb of repetitive sequences and 45,986 protein-coding genes were annotated. This genome assembly provides critical insights into A. chinensis’s genomic architecture and serves as a foundational resource for elucidating disease resistance mechanisms against Psa, while enabling comparative phylogenomic studies across the Actinidia genus.
Similar content being viewed by others

Background & Summary
Kiwifruit (Actinidia Lindl.), known as the “king of fruits” due to its exceptional phytochemical profile characterized by elevated ascorbate concentrations, potassium abundance, and dietary fiber content. This woody vine, native to China’s mountains, adapts to a wide range of habitats, including forest edges, roadside thickets, drainage ditches, and shrubby undergrowth. The genus comprises 54 taxonomically validated species exhibiting extensive cytogenetic variation with ploidy levels spanning diploid (2n = 2x) to octoploid (2n = 8x), while maintaining a conserved base chromosome number (x = 29)1.
Despite this genomic wealth, global commercial production remains constrained to four domesticated taxa: A. chinensis var. chinensis, A. chinensis var. deliciosa, A. eriantha, and A. arguta. According to 2024 FAO statistical data, the global kiwifruit production matrix encompasses 286,100 ha cultivation area yielding 4.29 million metric tons annually2. This agricultural success is threatened by pandemic outbreaks of bacterial canker caused by Pseudomonas syringae pv. actinidiae (Psa), which is a devastating disease that occurs in kiwifruit growing areas worldwide3. The pathogenic infection exhibits rapid proliferation and poses a persistent challenge for containment, primarily colonizing the plant’s meristematic tissues including emerging shoots, foliar structures, and cambial regions. This colonization manifests as necrotic branch cankers, chlorotic leaf lesions, and eventual systemic plant decline, severely compromising both fruit production and phytosanitary quality. Current phytoprotection strategies predominantly depend on cupric compounds and bacteriostatic agents, however, their protracted field application raises substantial ecological concerns regarding microbial resistance development and environmental persistence4,5.
Since the initial release of the kiwifruit draft reference genome (A. chinensis, 2n = 58) in 2013, subsequent genomic assemblies across Actinidia species have been progressively published, substantially propelling advancements in kiwifruit functional genomics research6. However, these assemblies have substantial gaps and unanchored sequences. The advent of long-read sequencing technologies, including Pacific Biosciences (PacBio) HiFi and Oxford Nanopore Technology (ONT) ultra-long platforms, has enabled precise resolution of complex genomic architectures and accurate assembly of highly repetitive chromosomal regions through their ultra-long sequencing reads7. To date, long-read sequencing technologies have enabled high-quality genome assemblies for multiple kiwifruit species, including A. chinensis, A. deliciosa, A. eriantha, A. arguta, A. latifolia, A. rufa, A. polygama, A. hemsleyana and A. zhejiangensis8,9,10,11,12,13,14,15,16,17,18,19,20. However, the lack of high-quality genome assemblies for canker-resistant kiwifruit cultivars, particularly the Psa-resistant A. chinensis var. chinensis ‘Guimi No.2’ (Accession No. 13-6-11, 2n = 58) discovered in Guizhou karst ecosystems, continues to impede comprehensive studies on disease resistance mechanisms and the functional characterization of critical resistance (R) genes.
In this study, we utilized short-read, PacBio HiFi long-read sequencing and Hi-C technology to generate a high-quality, chromosome-level assembly of A. chinensis var. chinensis ‘Guimi No.2’ genome. In conclusion, this genome provides valuable genetic resources for underlying the disease resistance mechanisms of the kiwifruit.
Methods
Materials collection and sequencing
Fresh young leaves used for genome sequencing were collected from the A. chinensis var. chinensis ‘Guimi No.2’ plant grown in the kiwifruit germplasm resource garden of Guizhou, China. A modified cetyltrimethyl ammonium bromide (CTAB) method was used for DNA extraction21. A second-generation sequencing library was constructed using X paired-end DNBSEQ-T7 libraries (MGI, Shenzhen, China), generating 80.60 Gb of raw sequencing data (PE150 reads). High-quality and purified genomic DNA samples were obtained, then, a SMRT cell sequencing library containing about 15–20 kb cut fragment was constructed and sequenced using PacBio sequel II sequencing platform. In total, 52.78 Gb raw data were obtained. A Hi-C library was established using the Illumina NovaSeq. 6000 platform (Illumina, San Diego, CA, USA) was used for sequencing. After the sequencing data of Hi-C was filtered, a total of 72.27 Gb raw data was obtained. Total RNA was isolated from a total of four tissues (root, stem, leaf, fruit) using the TRIzol reagent (Vazyme, China) for transcriptome sequencing. A cDNA library for transcriptome sequencing was constructed using the MGI Easy RNA Library Prep Kit (MGI, Shenzhen, China) with 350 bp insert sizes, and paired-end sequencing was performed on the DNBSEQ-T7 platform (MGI, Shenzhen, China), generating 25.96 Gb of raw sequencing data (PE150 reads) (Table 1).
Genome survey
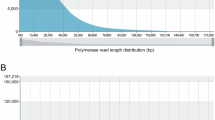
To estimate the genome size, the clean MGI sequencing reads were filtered with Fastp22 (v0.23.4) for subsequent k-mer analysis. Jellyfish23 (v2.3.0) was used to calculate the optimal k-mer and GenomeScope24 (v2.037) was used to estimate the genome size for corresponding k-mers. When k = 19, the genome size was about 634.80 Mb, and the heterozygosity was 0.91% (Fig. 1).

The characteristics of Guimi No.2 genome.
Chromosome-level genome assembly
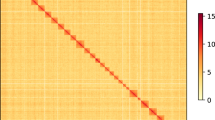
The genome was de novo assembled using Hifiasm25 (v0.24.0-r703) in Hi-C integrated assembly mode by combining PacBio HiFi long-read sequencing data with Hi-C chromatin interaction information. The primary assembly generated with default parameters was subsequently processed with purge_dups26 (v1.2.5) to eliminate redundancies. Chromosome-level scaffolding was performed using the HapHiC27 (v1.0.6) pipeline with species-specific parameterization (chromosome number set as 29). After manually checking and correcting in Juicebox28 (v2.1543), the pipeline finally generated a chromosome-level genome. Following the adjustment of chromosome coordinates, the Hi-C contact matrix heatmap was visualized with HiC-Pro29 and EndHiC30 (Fig. 2).

Hi-C contact matrix heatmap of Guimi No. 2 genome.
Based on the assembly pipeline, we obtained a genome size of 608.43 Mb with a contig N50 size of 20.70 Mb. The completeness of assembled genome was evaluated using BUSCO31 (v5.8.2) with the eudicots_odb10 dataset (comprising 2,326 single-copy orthologs), the analysis revealed that 98.40% (2,289/2,326) of conserved genes were fully detected (Complete BUSCOs, C), including 68.83% (1,601/2,326) as single-copy (S) and 29.57% (688/2,326) as duplicated (D), with 57 complete BUSCOs containing internal stop codons. 1.20% (28/2,326) of genes were fragmented (F), 0.38% (9/2,326) were missing (M), and 2.50% were excluded due to alignment issues (E) .
Genome annotation
EDTA32 (v2.2.2) pipeline was used to annotate repeat elements in the ‘Guimi No.2’ genome. A total of 251.15 Mb repeat sequences were identified, accounting for 41% of the entire genome. Gene structure annotation was performed using the BRAKER333 (v3.0.8) pipeline, which automates genome annotation by integrating transcriptome-based prediction (RNA-Seq data from root, stem, leaf, and fruit tissues generated in this study), homology-based prediction (A. chinensis, A. eriantha, A. arguta, A. zhejiangensis and A. melanandra), and ab initio prediction implemented through GeneMark-ETP34 (v1.02) and AUGUSTUS35 (v3.5.0). The pipeline utilized TSEBRA36 to merge evidence from RNA-Seq alignments (HISAT237 v2.2.1), protein homology, and ab initio predictions. Gene models were filtered to retain only those containing both start and stop codons and exceeding 100 nucleotides in length, yielding a final consensus set of 45,986 genes. Functional annotation was performed using emapper38 (v2.0.1) based on eggNOG orthology data.
The completeness of protein-coding genes in the assembled genome was evaluated using BUSCO (v5.8.2) with the eudicots_odb10 dataset (comprising 2,326 single-copy orthologs), the analysis revealed that 96.56% of conserved genes were fully detected (Complete BUSCOs, C), including 51.54% (1,199/2,326) as single-copy (S) and 45.01% (1,047/2,326) as duplicated (D). Only 0.94% (22/2,326) of genes were fragmented (F), while 2.49% (58/2,326) were missing (M). To show the characteristics of the Guimi No. 2 genome, we exhibited GC content, gene density, and repeats density (Fig. 3).

Circos plot of Guimi No. 2 genome. (A) Gene density, (B) GC content, (C) Repeat density, (D) Chromosome sizes.
Resistance gene analysis
The Psa-susceptible kiwifruit cultivar Hongyang10 (A. chinensis var. chinensis, 2n = 58) and the Psa-resistant cultivar Guimi No.2 were selected for comparative analysis of structural variations by MUMmer (v4.0.1, nucmer–mum)39. The structural variations identified included: 52 BRK (An insertion in the reference of unknown origin, that indicates no query sequence aligns to the sequence bounded by gap-start and gap-end, total length: 1,580,120 bp, accounting for 0.25% of the whole genome); 16,156 GAP (A gap between two mutually consistent ordered and oriented alignments, total length: 62,619,327 bp, 10.29% of the genome); 82 INV (The same as a relocation event, however both the ordering and orientation of the alignments is disrupted, total length: 2,187,148 bp, 0.35% of the genome); 217 JMP (A relocation event, where the consistent ordering of alignments is disrupted, total length: 5,104,265 bp, 0.83% of the genome); and 197 SEQ (A translocation event that requires jumping to a new query sequence in order to continue aligning to the reference, total length: 2,520,277 bp, 0.41% of the genome). Genome-wide identification of nucleotide-binding leucine-rich repeat (NLR) domains was performed using NLR-Annotator40 (v2.1b) with default parameters, revealed 252 and 282 putative NLR genes in Hongyang and Guimi No.2 (Fig. 4). Statistical analysis of the positional distribution of these SVs revealed that 205 SVs are located within the gene bodies and the 2-kb flanking regions upstream and downstream of 205 NLRs identified in Guimi No.2 (Supplementary Table 1).

Distribution of NLR genes along chromosomes in Guimi No. 2.
Data Records
The whole-genome sequencing raw data were deposited to the NCBI Sequence Read Archive with accession number SRR3279928541 (NGS-LEAF), SRR3279928642 (HIFI-LEAF), SRR3279928743 (HIC-LEAF), SRR3279928844 (RNA-LEAF), SRR3279928945 (RNA-FRUIT), SRR3279929046 (RNA-STEM), SRR3279929147 (RNA-ROOT). The assembled genome sequences have been deposited in the NCBI GenBank with the accession number JBMUMY000000000.148. The genome and annotation files were also deposited to the Figshare database49.
Technical Validation
The quality of the genome assembly was analyzed in the following aspects (Table 2): (1) The BUSCO score of 98.40%, 96.56% depicted the degree of completeness of the assembled genome and annotation. (2) 99.92% of PacBio HiFi reads were remapped to the assembled genome. (3) The quality value (QV) of the assembled genome was estimated using Merqury50 (v1.3), resulting in a value of 72.23 (long reads). (4) The LTR Assembly Index (LAI) of the assembled genome was estimated using LTR_FINDER_parallel51 (v1.07) and LTR_retriever52 (v2.9.5), resulting in a value of 10.10 which can be categorized as ‘Reference’ level. (5) Overall, these metrics indicate that the genome assembly of ‘Guimi No.2’ is of high quality and well-annotated.
Code availability
All software with their specific version used for data processing are clearly described in the methods section. If no specific variable or parameters are mentioned for a software, the default parameters were used.
References
Wu, Z. et al. Flora of China. Beijing: Science Press, 334–359 (2007).
FAO. FAOSTAT Online Database. http://www.fao.org/faostat/en/#data accessed September 2024 (2022).
Vanneste, J. Pseudomonas syringae pv. actinidiae (Psa): a threat to the New Zealand and global kiwifruit industry. N Z J Crop Hort Sci. 40, 265–7 (2012).
Wang, H. et al. Paenibacillus polymyxa YLC1: a promising antagonistic strain for biocontrol of Pseudomonas syringae pv. actinidiae, causing kiwifruit bacterial canker. Pest Manag Sci. 79, 4357–66 (2023).
Wang, Q. et al. Bioactivity and control efficacy of the novel antibiotic tetramycin against various kiwifruit diseases. Antibiotics. 10, 289 (2021).
Huang, S. et al. Draft genome of the kiwifruit Actinidia chinensis. Nat Commun 4, 2640 (2013).
Naish, M. et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science. 374, eabi7489 (2021).
Wang Y et al. Graph-Based Pangenome of Actinidia chinensis Reveals Structural Variations Mediating Fruit Degreening. Adv Sci (Weinh). 11(28), e2400322 (2024).
Yu X et al. Super pan-genome reveals extensive genomic variations associated with phenotypic divergence in Actinidia. Mol Hortic. 5(1), 4 (2025).
Yue J et al. Telomere-to-telomere and gap-free reference genome assembly of the kiwifruit Actinidia chinensis. Hortic Res. 10(2) (2022).
Han X et al. Two haplotype-resolved, gap-free genome assemblies for Actinidia latifolia and Actinidia chinensis shed light on the regulatory mechanisms of vitamin C and sucrose metabolism in kiwifruit. Mol Plant. 16(2), 452–470 (2023).
Xia, H. et al. Chromosome-scale genome assembly of a natural diploid kiwifruit (Actinidia chinensis var. deliciosa). Sci Data. 10, 92 (2023).
Liu Y et al. Chromosome-level genome of putative autohexaploid Actinidia deliciosa provides insights into polyploidisation and evolution. Plant J. 118(1), 73–89 (2024).
Wang Y et al. Telomere-to-telomere and haplotype-resolved genome of the kiwifruit Actinidia eriantha. Mol Hortic. 3(1), 4 (2023).
Yao X et al. The genome sequencing and comparative analysis of a wild kiwifruit Actinidia eriantha. Mol Hortic. 2(1), 13 (2022).
Liao, G. et al. A high-quality genome of Actinidia eriantha provides new insight into ascorbic acid regulation. J INTEGR AGR 22, 11 (2023).
Zhang F et al. Haplotype-resolved genome assembly provides insights into evolutionary history of the Actinidia arguta tetraploid. Mol Hortic. 4(1), 4 (2024).
Lu X et al. Genome assembly of autotetraploid Actinidia arguta highlights adaptive evolution and enables dissection of important economic traits. Plant Commun. 5(6), 100856 (2024).
Akagi, T. et al. Recurrent neo-sex chromosome evolution in kiwifruit. Nat Plants 9, 393–402 (2023).
Yu X et al. Genomic analyses reveal dead-end hybridization between two deeply divergent kiwifruit species rather than homoploid hybrid speciation. Plant J. 115(6), 1528–1543 (2023 Sep).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8 (1997).
Chen, S. F. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat Plants 10, 1184–1200 (2024).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101 (2016).
Servant, N. et al. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259 (2015).
Wang, S. et al. EndHiC: assemble large contigs into chromosome-level scaffolds using the Hi-C links from contig ends. BMC Bioinformatics 23, 528 (2022).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20, 275 (2019).
Gabriel, L. et al. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. Genome Res 34, 769–777 (2024).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics Bioinforma. 2, 1–14 (2020).
Stanke, M. et al. AUGUSTUS: A b initio prediction of alternative transcripts. Nucleic Acids Res. 34, 435–439 (2006).
Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22, 1–12 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 38, 5825–5829 (2021).
Marcais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Steuernagel, B. et al. The NLR-annotator tool enables annotation of the intracellular immune receptor repertoire. Plant Physiol. 183, 468–482 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32799285 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32799286 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32799287 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32799288 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32799289 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32799290 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32799291 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBMUMY000000000.1 (2025).
Figshare https://doi.org/10.6084/m9.figshare.28706651 (2025).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA 10, 48 (2019).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol 176, 1410–1422 (2018).
Acknowledgements
This work was supported by Guizhou provincial science and technology support program (Qiankehe [2023] 045), Guizhou key laboratory of molecular breeding for characteristic horticultural crops (Qiankehepingtai ZSYS [2025] 027), Guizhou provincial major scientific and technological program (Qiankehe [2024] 026), China Agriculture Research System (CARS-26).
Author information
Authors and Affiliations
Contributions
Jia Zhou and Dongmei Tang conceived and designed the project and the strategy. Weiming Zhong, Yong Qi, Binbin Shi, Qing Liu, Sheng Zhang and Nan Wang collected the samples. Jia Zhou assembled the genome. Jia Zhou and Feifei Wang wrote the original paper. Dongmei Tang and Qianming Zheng revised the original paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, J., Wang, F., Zhong, W. et al. A chromosome-level genome assembly of Guimi No. 2 (Actinidia chinensis). Sci Data 12, 1334 (2025). https://doi.org/10.1038/s41597-025-05593-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05593-6
