
Abstract
Electrophorus voltai, a South American electric eel, is renowned as the most powerful bioelectricity generator, capable of producing electric discharges reaching 860 volts. This remarkable ability positions it as an invaluable model for investigating the genetic, physiological, and developmental mechanisms driving electrogenesis in vertebrates. Here, we report a chromosome-level genome assembly of E. voltai, constructed using PacBio HiFi long reads and Hi-C scaffolding. The final assembly spans 666.91 Mb, with a contig N50 of 1.54 Mb and a scaffold N50 of 28.42 Mb, anchored onto 26 chromosomes. Genome annotation revealed 23,221 protein-coding genes, of which 22,306 genes were functionally annotated. Repetitive sequences account for 298.83 Mb of the genome, dominated by transposable elements. Additionally, we identified 225 miRNAs, 5,409 tRNAs, 288 rRNAs, and 535 snRNAs. This genome provides a foundational resource for comparative genomic studies of electric fish and facilitates investigations into the evolution of electrogenesis, electrocyte development, and bioelectric signal regulation in vertebrates.
Similar content being viewed by others

Background & Summary
Electric fishes have independently evolved electric discharges capabilities across multiple teleost lineages, most prominently within the order Gymnotiformes (South America)1. This unique adaptation has positioned electric fishes as valuable models for investigating convergent evolution, electroreception, and the diversification of ion channel functions1,2,3,4. Among them, the electric eel (genus: Electrophorus, family: Gymnotidae) represents one of the most prominent cases of high-voltage electrogenesis in vertebrates. The genus Electrophorus was long considered to consist of a single species, E. electricus. However, recent comprehensive taxonomic revision based on molecular and morphological analyses have reclassified Electrophorus genus into three distinct species: E. electricus, E. voltai, and E. varii5. Among them, E. voltai is distinguished by its ability to generate electric discharges reaching up to 860 volts, the highest voltage recorded in electric fishes. These high-voltage discharges fulfill multiple ecological functions, including predation, defense, navigation, and communication1,6. As a member of the order Gymnotiformes, E. voltai serves as an important model for unraveling the evolution and functional mechanisms of electric organ. Its unparalleled capacity for high-voltage discharge offers valuable insights into the genetic, physiological, and developmental foundations of biological electricity generation, shedding light on the evolutionary origins of bioelectrogenesis in vertebrates.
Despite its significance, no high-quality reference genome has been reported for E. voltai. This absence has hindered functional and comparative genomic studies in this species, limiting the identification of structural variants, regulatory elements, and conserved synteny critical to elucidating the molecular basis of electric organ development. Although existing genomic resources for electric fishes have improved, they remain relatively limited in taxonomic scope and quality. Many are restricted to draft assemblies or transcriptomic datasets from distantly related taxa, which lack the resolution needed to detect lineage-specific adaptations or interpret regulatory complexity3,7. The recent telomere-to-telomere assembly of E. electricus represents a significant advance8, yet additional high-quality genomes are still needed to enable robust comparative and evolutionary analyses. Furthermore, the evolutionary convergence of electrogenesis in distantly related lineages, including South American Gymnotiformes and African Mormyridae, highlights the necessity for species-specific genomic resources to dissect the distinct genetic architectures underlying similar electrogenic functions1,2,9. Thus, a high-quality genome for E. voltai is therefore indispensable, not only for elucidating species-specific adaptations in electrogenesis, but also for enabling robust cross-lineage comparisons and informing broader understanding of vertebrate organ evolution and physiological innovation.
To address this gap, we report a chromosome-level genome assembly of E. voltai, constructed using PacBio HiFi long-read and Hi-C sequencing. The assembled genome spans 666.91 Mb, with a scaffold N50 of 28.42 Mb and contig N50 of 1.54 Mb, anchored onto 26 chromosomes. BUSCO assessment revealed that 95.80% of the expected conserved orthologs were complete in the genome assembly, and gene prediction identified 23,221 protein-coding genes, of which 22,306 (96.06%) were functionally annotated. This high-quality genomic resource provides a robust foundation for future studies of the molecular basis of electric signal production, electrocyte development, and functional adaptations in electric fishes. It also enables comprehensive investigations into genome architecture, gene family evolution, and regulatory networks underpinning vertebrate electrogenesis, offering new avenues to study the evolutionary innovation of bioelectricity.
Methods
Ethics statement
The experimental protocols involving animals were reviewed and approved by the Laboratory Animal Ethics Committee of the Pearl River Fisheries Research Institute, (PRFRI), Chinese Academy of Fishery Sciences (CAFS), China (License No. LAEC-PRFRI-2020-11-17).
Sample collection and identification
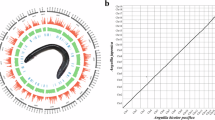
Electric eel sample was collected from Guangzhou Lanhai Marine Technology Co., Ltd (Guangzhou City, Guangdong Province, China), and identified as E. voltai through COI barcoding. The obtained COI sequence was compared against all available COI sequences of the genus Electrophorus in NCBI GenBank. Phylogenetic analysis showed that our sequence clustered unambiguously with E. voltai reference sequences, clearly distinguishing it from E. electricus and E. varii (Figure S1). The sampled E. voltai individual was confirmed to be male based on anatomical examination, with a total length of 1.15 meters (Fig. 1a). E. voltai possesses three distinct electric organs: the main electric organ (main EO), Hunter’s organ, and Sach’s organ. The main EO is primarily responsible for generating high-voltage discharges3 and was the tissue sampled for both genome sequencing and RNA sequencing in this study.

Specimen photograph and Hi-C chromatin interaction heatmap of Electrophorus voltai. (a) Photograph of the E. voltai specimen. (b) Heatmap of chromatin interactions across the E. voltai genome based on Hi-C data. Genomic coordinates (in megabases) are displayed along both axes, while interaction intensity is represented by color shading. Darker reds indicate higher interaction frequencies. The color bar on the right denotes interaction strength.
Genome sequencing
Genomic DNA was extracted from the main electric organ (EO) of E. voltai using the TIANamp Genomic DNA Kit (TIANGEN, Beijing, China). DNA integrity was verified by gel electrophoresis (1.0% agarose), concentration was measured using a NanoDrop 2000 spectrometer (Thermo Scientific, Waltham, MA, USA). Qualified genomic DNA was fragmented to 300–500 bp using a Covaris ultrasonicator (Covaris Inc., Woburn, MA, USA), followed by end-repair, A-tailing, adapter ligation and PCR amplification. A short-insert (350 bp) paired-end library was constructed and sequenced on the MGIseq. 2000 platform (MGI Tech Co. Ltd., Shenzhen, Guangdong, China), yielding approximately 40.1 Gb of 150 bp paired-end reads (Table 1). For long-read sequencing, high-fidelity (HiFi) libraries were constructed following PacBio’s standard protocol (Pacific Biosciences, Menlo Park, CA, USA), and sequenced on the PacBio Sequel II system, generating approximately 13.8 Gb of HiFi reads. Additionally, for chromosome-level scaffolding, Hi-C libraries were prepared from EO-derived genomic DNA that was cross-linked with formaldehyde and digested using the MboI restriction enzyme. The Hi-C libraries were sequenced on the MGISEQ-2000 platform, producing approximately 46.3 Gb of data (~81X coverage).
Transcriptome sequencing
Total RNA was extracted from the main EO with a TRIzol kit (Invitrogen, Carlsbad, CA, USA), and mRNAs were then isolated and purified from the sample with an Oligotex mRNA Midi Kit (Qiagen GmbH, Hilden, Germany). RNA concentration and purity were measured by a Nanodrop spectrophotometer. RNA integrity was verified with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). A transcriptome library was constructed by using NEBNext Ultra RNA Library Prep Kit (NEB, Ipswich, MA, USA) according to the manufacturer’s protocol, which was then sequenced on a MGIseq 2000 platform following the 150-bp paired-end protocol.
Genome survey and assembly
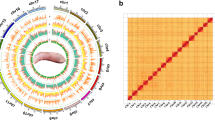
Prior to genome assembly, k-mer frequency analysis was performed using short-read data generated on the MGIseq 2000 platform to estimate the genome characteristics of E. voltai, including genome size, heterozygosity, GC content, and repeat content. K-mer counting was conducted with Jellyfish v2.010, and the 17-mer frequency distribution was analyzed (Figure S2). The genome size was estimated to be approximately 773.91 Mb, with a heterozygosity ratio of 0.89%, GC content of 39.9% and repetitive sequences accounting for 51.13% of the genome. The genome of E. voltai was initially assembled into contigs using PacBio HiFi reads using hifiasm v0.14-r31211, generating 1,183 contigs with a total length of 692.97 Mb and a contig N50 of 1.47 Mb. Further, we also integrated Hi-C data to obtain high quality de novo assembly at the chromosome level. Quality control of raw Hi-C reads and the generation of valid paired-end interactions were performed using Juicer v1.5 (RRID:SCR_017226)12. Scaffolding was conducted using the 3D-DNA pipeline v18092213, anchoring 87.01% of the assembled contigs onto 26 chromosomes based on Hi-C interaction data (Fig. 1b, Table 2). The resulting chromosome number is consistent with the karyotypic data previously reported for Electrophorus electricus14,15. The final genome assembly spanned 666.91 Mb, with a scaffold N50 of 28.42 Mb and a contig N50 of 1.54 Mb (Fig. 2, Table 3).

Circos plot illustrating the genomic landscape of E. voltai. From the outermost to innermost rings: (a) chromosome layout (Chr1-Chr26), (b) distribution of gene density (0–100), (c) repeat element coverage (0–55), (d) localization of non-coding RNAs, including rRNAs (black), snRNAs (red), tRNAs (green), and miRNAs (blue), and (e) GC ratio (30%–55%).
Repeat identification
Repetitive elements in the E. voltai genome were identified using a combination of ab initio and homology-based approaches. For ab initio prediction, RepeatModeler v1.0.1116 and LTR-FINDER v1.0.517 were used to construct a de novo repeat library, which was subsequently applied to the genome using RepeatMasker v4.0.918. Tandem repeats were detected with Tandem Repeats Finder (TRF) v4.0919. For homology-based detection, genome sequences were aligned to the RepBase database (http://www.girinst.org/repbase) using both RepeatMasker v4.0.918 and RepeatProteinMask v4.0.918. By integrating the results from both ab initio and homology-based methods, a total of 298.83 Mb of repetitive sequences were identified, accounting for 43.09% of the genome (Table 4). Among these, transposable elements (TEs) constituted 37.35% of the genome.
Gene annotation
Gene prediction for E. voltai was conducted using an integrative approach combining homology-based, transcriptome-based, and ab initio methods. For homology-based annotation, protein sequences from four phylogenetically related species were retrieved from NCBI, including E. electricus1, Ictalurus punctatus20, Silurus meridionalis21, and Danio rerio22. These sequences were aligned to the E. voltai genome using BLAST v2.11.0+23 with an e-value cutoff of 1e–5. Gene structures were further refined using Exonerate v2.4.024. For transcriptome-based prediction, raw RNA-seq reads were filtered using SOAPnuke v2.1.025 with parameters -lowQual = 20 -nRate = 0.005 -qualRate = 0.5. Clean reads were assembled de novo with Trinity v2.8.526, aligned to the assembled genome using HISAT2 v2.2.127, and assembled into transcripts with StringTie v2.1.728. Ab initio gene prediction was performed with Augustus v3.429 and Genscan v1.030. Gene models derived from the three strategies were integrated into a non-redundant reference gene set using MAKER v3.01.0331, followed by refinement with PASA v2.4.132. By integrating results from all three annotation strategies, we predicted 23,221 protein-coding genes (Table 5). Functional annotation was performed using DIAMOND v2.0.733 by aligning the predicted proteins against the InterPro34, SwissProt, TrEMBL35, NR, GO36, and KEGG37 databases. As a result, 96.06% (22,306) of the predicted genes received functional annotations (Table 6).
Non-coding RNA annotation
Non-coding RNAs (ncRNAs) were annotated using a combination of tools appropriate for each class of ncRNA. Transfer RNAs (tRNAs) were identified using tRNAscan-SE v2.0.938, while ribosomal RNAs (rRNAs) were predicted with RNAmmer v1.239. For the identification of microRNAs (miRNAs) and small nuclear RNAs (snRNAs), we employed INFERNAL v1.1.440 as implemented in the Rfam database41. As a result, a total of 7,279 ncRNA genes were annotated in the E. voltai genome, including 225 miRNAs, 5,409 tRNAs, 576 rRNAs and 1,069 snRNAs (Table 7).
Data Records
All raw sequencing data generated in this study have been deposited in the NCBI SRA database under the BioProject accession number PRJNA946944. Specifically, short-read data (SRR33386860)42, PacBio HiFi reads (SRR33386859)43, Hi-C reads (SRR33386858)44, and RNA-seq reads (SRR33386857)45 are publicly available. The final chromosome-level genome assembly has been deposited in the GenBank database under accession number JAROKS00000000046, and the corresponding genome annotation files are available on Figshare47.
Technical Validation
The E. voltai genome assembly was assessed for completeness and annotation quality through four independent methods. First, assessment of completeness using BUSCO v5.2.248 demonstrated high completeness levels for both the genome assembly (95.80%) and the predicted gene set (95.50%) (Table 3). Second, 99.94% of the PacBio HiFi clean reads were successfully mapped back to the assembly using minimap2 v2.2149, indicating high assembly accuracy. Third, to assess the accuracy and reliability of gene prediction, we compared the distributions of gene length, coding sequence (CDS) length, exon length, and intron length between E. voltai and four phylogenetically related species (E. electricus, I. punctatus, S. meridionalis, and D. rerio). The consistent distribution patterns across these species further validated the quality of the annotated gene dataset for E. voltai (Fig. 3). Fourth, we conducted a genomic collinearity analysis between E. voltai and E. electricus using the jcvi toolkit (Python version of MCScan50; https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version)). The analysis revealed a high degree of one-to-one correspondence at the chromosomal level, with only minor structural differences detected on chromosomes 4, 5, 8, 9, 14, 16, and 17 (Fig. 4). We also confirmed that key genes associated with electrogenesis, including voltage-gated sodium channel genes (scn4aa, scn4ab), acetylcholinesterase (ache), ATPase subunit genes (atp1a2, atp1b1b), and other related genes, are accurately annotated in the assembly. Among these, scn4aa is reported to play a key role in high-voltage generation in electric fishes and is particularly highly expressed in the electric organ1,2,3,4,9. Together, these results support the quality and reliability of the genome and its utility for future comparative and evolutionary studies.

Comparative analysis of genomic features across E. voltai and four closely related species. (a) Distribution of gene lengths, highlighting interspecific differences in overall gene architecture. (b) Comparison of coding sequence (CDS) lengths, illustrating variation in protein-coding region sizes. (c) Exon length distributions across species, showing differences in exon structural composition. (d) Intron length comparisons, indicating species-specific variation in non-coding genomic regions.

Chromosomal synteny between E. voltai and E. electricus. Each colored block represents a chromosome.
Code availability
In this study, all bioinformatic tools and software were used according to their respective manuals, with default parameters applied unless otherwise specified. The Methods section provides details of the software versions, and the codes or parameters used. No custom scripts were employed.
References
Gallant, J. R. et al. Genomic basis for the convergent evolution of electric organs. Science 344, 1522–1525 (2014).
LaPotin, S. et al. Divergent cis-regulatory evolution underlies the convergent loss of sodium channel expression in electric fish. Sci Adv 8, eabm2970 (2022).
Traeger, L. L. et al. Unique patterns of transcript and miRNA expression in the South American strong voltage electric eel (Electrophorus electricus). BMC Genomics 16, 1–14 (2015).
Zakon, H. H. Adaptive evolution of voltage-gated sodium channels: the first 800 million years. Proc. Natl. Acad. Sci. 109, 10619–10625 (2012).
De Santana, C. D. et al. Unexpected species diversity in electric eels with a description of the strongest living bioelectricity generator. Nat. Commun. 10, 4000 (2019).
Güth, R., Pinch, M. & Unguez, G. A. Mechanisms of muscle gene regulation in the electric organ of Sternopygus macrurus. J. Exp. Biol. 216, 2469–2477 (2013).
Wang, Y. & Yang, L. Genomic evidence for convergent molecular adaptation in electric fishes. Genome Biol. Evol. 13, evab038 (2021).
Qi, Z. et al. Telomere-to-telomere genome assembly of Electrophorus electricus provides insights into the evolution of electric eels. GigaScience 14, giaf024 (2025).
Zakon, H. H. et al. Sodium channel genes and the evolution of diversity in communication signals of electric fishes: convergent molecular evolution. Proc. Natl. Acad. Sci. 103, 3675–3680 (2006).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Cardoso, A. L. et al. Chromosomal variability between populations of electrophorus electricus Gill, 1864 (Pisces: Gymnotiformes: Gymnotidae). Zebrafish 12, 440–447 (2015).
Fonteles, S. et al. Cytogenetic characterization of the strongly electric Amazonian eel, Electrophorus electricus (Teleostei, Gymnotiformes), from the Brazilian rivers Amazon and Araguaia. Genet Mol Biol. 31, 227–230 (2008).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 5, 4.10. 1–4.10. 14 (2004).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Liu, Z. et al. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 7, 11757 (2016).
Zheng, S. et al. Chromosome‐level assembly of southern catfish (silurus meridionalis) provides insights into visual adaptation to nocturnal and benthic lifestyles. Mol Ecol Resour. 21, 1575–1592 (2021).
Howe, K. et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 (2013).
Altschul, S. F. et al. Basic local alignment search tool. J Mol Biol. 215, 403–410 (1990).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 6, 1–11 (2005).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, gix120 (2018).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29, 644–652 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 12, 357–360 (2015).
Pertea, M. et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 11, 1650–1667 (2016).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 268, 78–94 (1997).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 1–14 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 18, 366–368 (2021).
Finn, R. D. et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 27, 49–54 (1999).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat Genet. 25, 25–29 (2000).
Kanehisa, M. et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Chan, P. P. et al. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33386860 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33386859 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33386858 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33386857 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:JAROKS000000000 (2025).
Mu, X. D. The genome annotation of Electrophorus voltai. figshare https://doi.org/10.6084/m9.figshare.28891460 (2025).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49 (2012).
Acknowledgements
This work was supported by the Rural Revitalization Strategy Special Provincial Organization and Implementation Project Funds (2023SBH00001), the Guangdong Provincial Special Fund for Modern Agriculture Industry Technology Innovation Teams (2024CXTD26), China-ASEAN Maritime Cooperation Fund (CAMC-2018F), National Freshwater Genetic Resource Center (FGRC18537).
Author information
Authors and Affiliations
Contributions
Yuanyuan Wang: Conceptualization; data curation and analysis; investigation; writing (original draft, review and editing). Yi Liu: Sample collection, data curation; methodology; writing (review and editing). Yexin Yang: Sample collection, resources, writing (review and editing). Chao Bian, Chao Liu: Sample collection, writing (review and editing). Yang Zhang: Conceptualization; sample collection, data curation; resources, writing (review and editing). Xidong Mu: Conceptualization; data curation; funding acquisition; resources; supervision; writing (review and editing).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Liu, Y., Yang, Y. et al. A chromosome-level genome assembly of Electrophorus voltai, a species of electric eel. Sci Data 12, 1367 (2025). https://doi.org/10.1038/s41597-025-05720-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05720-3
