
Abstract
Camellia fascicularis is a golden camellia with important ornamental and economic value. In 2010, it was listed as a species with extremely small populations (PSESP) in urgent need of rescue and protection in Yunnan, China, due to the fragmented habitat, extremely small quantity, and significant human interference. However, how to protect and manage at the genomic level still faces challenges. Thus, a high-quality chromosome-level genome of C. fascicularis was assembled by combining the PacBio HiFi sequencing and high-throughput chromosome conformation capture (Hi-C) sequencing. The final assembled genome size is 5.65 Gb, comprising two complete haplotypes, with a contig N50 of 115.51 Mb (57 gaps) and a scaffold N50 of 191.08 Mb; 99.67% of contigs are anchored to 30 pseudo-chromosomes(15 per haplotype), and all chromosomal ends contain telomeric characteristic motifs (TTTAGGG)n. In addition, the assembled genome comprises a total of 85,316 predicted coding genes, all of which have been functionally annotated. The first report on the genome of C. fascicularis will lay a solid foundation for future research on its protective genomics.
Similar content being viewed by others


Background & Summary
There are more than 1200 camellia species in the world, with the majority being red flowers and only a few number (83 species) of golden flowers (https://camellia.iflora.cn/)1, which belongs to the Camellia sect. Chrysantha. These golden camellias are evergreen shrubs or small trees, often praised as the “Queen of Camellias”. However, due to the serious threat, C. fascicularis has been listed as a second-class key protected wild plants in China2. The golden-flower camellias are precious resources that combine ornamental, medicinal, and edible purposes. In addition, they are valuable resources for studying the color formation and breeding the yellow-flower camellia cultivars1,3.
According to the IUCN evaluation criteria, C. fascicularis was categorized as critically endangered (CR) on the China biodiversity red list higher plants of 2020. It was also listed as one of the 62 and 101 target species in the Yunnan provincial conservation action plan for plants with extremely small populations (PSESP) in 2010 and 2021 respectively4. In addition to its high ornamental value in gardens, it is also an edible and medicinal plant, with its leaves rich in nutritional and health-promoting compounds. These leaves have antioxidant, anti-inflammatory, and other beneficial physiological activities5,6. In recent years, despite intensified conservation efforts for C. fascicularis, limitations in natural regeneration and genetic rescue persist. As a flagship PSESP endemic to southeastern Yunnan, its successful genomic conservation would establish a model for rescuing co-occurring endangered species in this biodiversity hotspot7,8,9,10. Therefore, developing genomic-level strategies is critical to enable effective protection mechanisms for this species and other PSESPs.
Plant genome sequencing has developed rapidly in the past 20 years, and by early December 2024, over 1,500 genome sequences of higher plant taxa have been published11. Sequencing genomes can provide insights and evidence for a better understanding of plant genome biology and evolution12,13. Although the genomes of so many plant species have been studied, only a few studies have sequenced the genomes of threatened plant species (such as Anisodus tanguticus14, Firmiana kwangsiensis15, Zanthoxylum nitidum16, C. nitidissima17, Rhododendron griersonianum18 and Magnolia sinica19), in order to focus on their conservation.
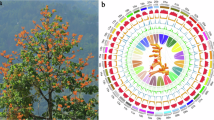
The de novo assembly of plant genomes is pivotal for advancing our understanding of plant evolution processes, ornamental plants breeding and conservation efforts15,20. In this study, we generated a high-quality chromosomal-level genome assembly of C. fascicularis (Fig. 1) based on PacBio HiFi reads (89.22 Gb) and Hi-C reads (249.45 Gb) (Fig. 2; Supplementary Tables 1, 2). The final assembly (~5.65 Gb) consisted of two complete haplotypes: haplotype A (~2.89 Gb) and haplotype B (~2.76 Gb), with contig N50 lengths of 196.04 Mb and 104.27 Mb, respectively (Table 1). The final genome assembly, comprising two haplotypes, was anchored onto 30 pseudochromosomes (15 per haplotype), with 99.67% of the assembled sequences successfully placed, as shown in Fig. 2 and Table 1. Telomeric repeat motifs (TTTAGGG)n were identified at the ends of all chromosomes, and the scaffold N50 reached 191.08 Mb. In addition, a total of 7,943,983 repetitive sequences were identified, with a total length of 3,853,635,256 bp, accounting for 68.21% of the genome. Among them, the most abundant are long terminal repeat sequences (LTRs), with a total of 4,349,137 elements and a cumulative length of 3,162,073,259 bp, accounting for 55.97% of the entire genome (Table 2; Supplementary Table 3). The genome of C. fascicularis contains a total of 88,796 genes, including 85,316 protein coding genes and 3,480 non-coding genes (Table 3). A total of 85,316 protein coding genes were functionally annotated (Table 4). This study obtained a high-quality genome of C. fascicularis, which is of being great significance for the conservation of this species.

Morphological Characteristics of C. fascicularis. (a,b) Flowers. (c) Seedlings. (d) Stems. (e) Leaves. (f) Roots of Seedlings.

The genomic landscape of 30 pseudo-chromosomes of C. fascicularis (labeled as Chr01-15[a, b]). Circles from outside to inside: (a) chromosome length, (b) density of Class I transposable elements (TEs), including long terminal repeats (LTRs) and long/short dispersed elements, (c) density of Class II TEs (DNA and Helitrons), (d) density of coding genes (mRNA), (e) proportion of tandem repeat sequences, (f) GC content, and (g) synteny blocks.
Methods
Plant material
For genomic sequencing, we collected fresh leaf samples from adult healthy plants of C. fascicularis in the wild from Hekou County, Yunnan Province (103°58′E, 22°52′N) (Fig. 1). In addition to the leaf samples, the flowers, roots, and young stem samples were also collected for full-length transcriptome sequencing. After collection, the samples were immediately wrapped in aluminum foil and quickly frozen in liquid nitrogen, then stored at −80 °C until further processing.
Genomic DNA extraction and sequencing
Genome DNA sequencing was performed using different sequencing platforms simultaneously to ensure accurate assembly. The specific steps are as follows:
High-quality genomic DNA was isolated from the fresh leaves using the CTAB method, and the DNA quality and concentration were tested by 0.75% agarose gel electrophoresis, NanoDrop One spectrophotometer (Thermo Fisher Scientific) and Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA).
The libraries used for single-molecule real-time (SMRT) Pacific Biosciences (PacBio) genome sequencing were constructed according to standard protocols. A library with a DNA-fragment insert size of ~15 kb was prepared from 3 μg of high-quality genomic DNA and placed into the Revio system (Pacbio, USA) sequencer for sequencing. A total of 89.22 Gb (~5.06 M reads) of HiFi sequencing data was obtained, which was used for the subsequent genome assembly of C. fascicularis (Supplementary Table 1).
High-quality gDNA was randomly fragmented by ultrasonic oscillation (Covaris, USA) and used for DNBSEQ-T7 (BGI Inc., China) short-read sequencing. To increase continuity of the genome, the gDNA was used to construct the Hi-C libraries according to the standard library preparation protocol21. The Hi-C libraries were enriched, A-tailed, and then subjected to PCR amplification (12–14 cycles). Subsequently, the Hi-C libraries were sequenced using the DNBSEQ-T7 platform, operating in PE150 mode. Approximately 249.45 Gb (~1663 M reads) of Hi-C data were generated for subsequent pseudochromosome assembly (Supplementary Table 2). Hi-C data were integrated with the PacBio assembly to resolve chromosomal structures.
RNA extraction and sequencing
For transcriptome sequencing, fresh tissue samples including stems, young and mature leaves, roots, and petals were collected from C. fascicularis and immediately frozen in liquid nitrogen. Total RNA was subsequently extracted using the TRIzol® Reagent (Invitroge). The concentration and quality of RNA were assessed using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and a Bioanalyzer 2,100 system (Agilent Technologies, CA, USA). High-quality RNA from different tissues was mixed in equal amounts and used to construct the cDNA libraries according to the manufacturer’s instructions. Libraries were then loaded onto an R9.4 sequencing chip and sequenced on a PromethION sequencer (Oxford Nanopore Technologies, UK). Finally, a total of 17.25 Gb (~13.84 M reads) of full-length RNA-seq data was obtained (Supplementary Table 4).
De novo genome assembly
First, contigs were assembled from PacBio HiFi reads using hifiasm (v0.19.8-r602), with haplotype assembly selected for subsequent analysis22. Next, Hi-C reads were aligned to the assembled contigs using Juicer23, followed by preliminary Hi-C-assisted chromosome scaffolding and anchoring with 3D-DNA24. The chromosome segmentation boundaries and assembly errors were manually checked and adjusted using Juicebox25. After manual review, the final chromosome framework and scattered sequences were generated. Additionally, the chloroplast and mitochondrial genomes were assembled using the GetOrganelle toolkit26.
To further optimize the assembly, the quarTeT software was used to fill in missing gaps based on the HiFi reads, thereby improving the completeness of the assembly. Additionally, by aligning the HiFi reads to the regions near the chromosome telomeres, the terminal sequences were assembled using hifiasm, and the chromosomes were extended to recover the telomere sequences as completely as possible. To remove redundancy and foreign contamination, redundans was employed to align scattered contigs with chromosomes and organelle genomes, eliminating redundant sequences, particularly the large fragments that might arise from organelle genomes and rDNA27. Finally, low-coverage fragments or haplotigs were identified and removed to ensure the purity and accuracy of the assembled data.
Finally, based on the reference tea28 genome, the chromosomes were numbered as chr01-15[ab], where “a” and “b” do not represent sequences from the same parent; the “a” set was chosen as the primary assembly result. All chromosome sequences were adjusted to ensure they were oriented in the same direction. According to the assembly results (Supplementary Table 5), a total of 30 chromosomes were identified, accounting for 99.67% of the total genome length. Chromosomes 1b, 2b, 8a, 9b, and 14a were fully assembled without gaps, including the successful assembly of their terminal sequences with chromosomes 1b, 2b, 8a, 9b, and 14a showing the best assembly quality (Supplementary Table 6).
Genome annotation
Repetitive sequence annotation
Several different methods were employed to annotate the repetitive sequences. First, transposable elements were identified de novo using EDTA29 (parameters -sensitive 1-anno 1) to generate a TE library. Then, RepeatMasker (http://www.repeatmasker.org/RepeatMasker/) was used to identify repetitive regions in the genome. A total of 3.85 Gb (68.21%) of the assembled sequences were annotated as TEs, with LTR (55.97%) and TIR (7.62%) being the two most abundant TE superfamilies (Fig. 2; Table 2; Supplementary Table 3).
Protein-coding gene annotation
For gene annotation, public protein sequences from C. sinensis, Diospyros lotus, Rhododendron ovatum, Gilia yorkii, Vitellaria paradoxa, Aegiceras corniculatum, Coffea canephora, Solanum lycopersicum, Cornus controversa, Vitis vinifera, and Arabidopsis thaliana were collected. A total of 339,202 non-redundant protein sequences were used as homologous protein evidence for gene annotation.
For comprehensive genome annotation, RNA-seq data were first aligned to the genome using minimap230, followed by transcript assembly with StringTie31 (Supplementary Table 7). Subsequently, the PASA pipeline32 was used for gene structure annotation of the transcripts, and full-length genes were identified through reference protein alignment. Based on the full-length gene set, AUGUSTUS33 and SNAP34 were trained to optimize the prediction models. In the MAKER235 pipeline, de novo predictions, transcript evidence, and homologous protein evidence were integrated. Repetitive regions were masked using RepeatMasker, and de novo (ab initio) gene predictions were made using AUGUSTUS33 and SNAP34. Meanwhile, the transcripts and protein sequences were aligned to the genome using BLASTN and BLASTX, and the alignment results were further optimized with Exonerate36, generating hint files to integrate the gene models. Since the annotation accuracy of the MAKER pipeline is relatively low37, the annotations from MAKER and PASA were further integrated using EVidenceModeler38 (EVM) to generate a consistent gene annotation. To avoid introducing TE coding regions, TE protein domains on the genome were identified using TEsorter39, and these regions were masked using EVM.
Non-coding RNA annotation
For non-coding RNA annotation, tRNA genes were annotated using tRNAScan-SE40, rRNA genes were annotated using barrnap (https://github.com/tseemann/barrnap/), and various non-coding RNAs, including miRNA and snRNA, were annotated through RfamScan. All annotations were merged, with redundant annotations removed (coding genes were prioritized, and overlapping genes were excluded). Finally, both coding and non-coding genes were uniformly named. In the end, all the annotation results were merged and redundancies were removed to get the complete set of genes.
Functional annotation
To further annotate the functions of protein-coding genes, three strategies were employed: (1) eggNOG-mapper41 annotation: Gene functions were annotated by comparing them with the eggNOG homologous gene database, including GO, KEGG, and other categories. (2) Sequence similarity search: DIAMOND42 was used to align protein sequences to protein databases (e.g., Swiss_Prot, TrEMBL, NR, and the Arabidopsis database) to identify the best matches for genes. The alignment criteria were an identity percentage greater than 30% and an E-value less than 1e-5. (3) Domain similarity search: InterProScan43 was used to compare with sub-databases within InterPro, such as PRINTS, Pfam, SMART, PANTHER, and CDD, to obtain conserved amino acid sequences, motifs, and domains of the proteins.)
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive44 in National Genomics Data Center45, China National Center for Bioinformation Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA024719)46 that are publicly accessible. The genome assembly and annotation data can also be accessed at Genome Warehouse(GWH) using the accession number GWHFSQE00000000.147. The genome assembly has also been submitted to the National Center for Biotechnology Information (NCBI) under the accession numbers GCA_051225895.1(CfHapA)48 and GCA_051225905.1 (CfHapB)49.
Technical Validation
Evaluation of the assembled genomes
In the genome assembly of C. fascicularis, we assembled two complete haplotypes with a total size of 5.65 Gb, including two haplotypes (Fig. 2; Supplementary Tables 1 and 2). The scaffold N50 was 191.08 Mb, indicating good continuity. The N50 of the contigs reached 115.51 Mb, with 57 gaps, and these contigs were anchored to 30 pseudochromosomes, with an anchoring rate of 99.67%. Additionally, the telomeric sequence (TTTAGGG)n was detected at the ends of the most chromosomes, and the 18S and 5S rDNA arrays were detected on chromosomes 7, 8, 11, and 13 (Fig. 4). All chromosomes contained a highly tandem repeat sequence (designated as cent2), with the unit sequence being AGAATTTACTGGGAATTTACTGAGTAATTTACTGAGAATTTACTG (Fig. 4). This sequence may be related to centromere function. Its sequence characteristics and distribution provide important clues for studying chromosome segregation mechanisms, centromere function, and chromosome evolution.
To further validate the assembly quality, the third-generation sequencing data (HiFi reads) and (Iso seq) were compared with the genome using bwa27 and minimap230, respectively (Supplementary Table 8). HiFi reads mapped to 99.64% of the assembly with ≥5 × coverage, ensuring minimal assembly gaps. The Hi-C interaction map showed a strong intrachromosomal interactive signal along the diagonal. The BUSCO (Benchmarking Universal Single-Copy Orthologs) assessment showed high completeness of the core genes (Supplementary Tables 9, and 10). A total of 99.13% of the core genes (including both single-copy and multi-copy genes) were successfully assembled, with a gene missing rate of 0.43%. Additionally, there were no significant heterozygous peaks in the BUSCO core single-copy and multi-copy gene regions, and their distribution was consistent (Fig. 3a,b). The Hi-C interaction heatmaps generated by Juicer show high resolution for each chromosome (Fig. 3c), with no obvious noise observed outside the diagonals, further supporting the quality of the assembly. Additionally, no anomalies were observed between each pair of homologous chromosomes (Fig. 3d), indicating no obvious switch errors.

Distribution of genome-wide (a) and BUSCO core gene regions (b) coverage depth evaluated using HiFi data; Hi-C interaction heatmaps for haplotypes A and B, depicting interactions for (c) reads with mapping quality of 0 or higher (including duplicates) and (d) mapping quality of 1 or higher (excluding duplicates). The color scale represents interaction intensity, with yellow signifying weak and red indicating strong interactions.

Profiles of repetitive sequence distribution across the chromosomes of C. fascicularis, highlighting telomeric TTTAGGG sequences, tandem repeats, 18S rDNA, and 5S rDNA. The vertical scale denotes the frequency of repetitive sequences in 20 kb segments. Black triangles signify the positions of gaps.
In this study, the assembled genome is characterized by minimal gaps, high telomere sequence integrity, elevated mapping rates, exceptional completeness, absence of redundancy, and high-resolution Hi-C interaction heatmaps, collectively demonstrating the superior quality of the genome assembly.
Evaluation of the gene annotation
The integrated annotation of proteins was evaluated using BUSCO, and the results are presented in Supplementary Tables 9, 10. The evaluation indicated that the complete core genes coverage was 99.44%, including 1.98% single-copy genes and 97.46% duplicated genes. The annotation was further characterized by a minimal presence of fragmented genes (0.31%) and missing genes (0.25%), collectively representing high-quality annotation.
Code availability
All commands and pipelines used were performed according to the manuals or protocols of the tools used in this study. The softwares and tools used are publicly accessible, with the version and parameters specified in the methods section. If no detailed parameters were mentioned, default parameters were used. No custom code was used in this study.
References
Feng, Y., Li, J., Yin, H., Shen, J. & Liu, W. Multi-omics analysis revealed the mechanism underlying flavonol biosynthesis during petal color formation in Camellia Nitidissima. BMC Plant Biology 24, 847, https://doi.org/10.1186/s12870-024-05332-w (2024).
Zhou, X. et al. De novo assembly of the Camellia nitidissima transcriptome reveals key genes of flower pigment biosynthesis. Frontiers in plant science 8, 1545, https://doi.org/10.3389/fpls.2017.01545 (2017).
Nabavi, S. M. et al. Flavonoid biosynthetic pathways in plants: Versatile targets for metabolic engineering. Biotechnology advances 38, 107316, https://doi.org/10.1016/j.biotechadv.2018.11.005 (2020).
Shi, Z. et al. UHPLC Q-Exactive Orbitrap MS based metabolomics and biological activities of Camellia fascicularis from different geographical regions. Industrial Crops and Products 213, 118432, https://doi.org/10.1016/j.indcrop.2024.118432 (2024).
Peng, X. et al. Extraction, characterization, antioxidant and anti-tumor activities of polysaccharides from Camellia fascicularis leaves. International Journal of Biological Macromolecules 222, 373–384, https://doi.org/10.1016/j.ijbiomac.2022.09.176 (2022).
Yu, S. et al. Identification of Chalcone Isomerase Family genes and roles of CnCHI4 in Flavonoid metabolism in Camellia Nitidissima. Biomolecules 13, 41, https://doi.org/10.3390/biom13010041 (2022).
Ma, Y. et al. Conserving plant species with extremely small populations (PSESP) in China. Biodiversity and Conservation 22, 803–809, https://doi.org/10.1007/s10531-013-0434-3 (2013).
Sun, W., Yang, J. & Dao, Z. Study and Conservation of Plant Species with Extremely Small Populations (PSESP) in Yunnan Province, China. Beijing: Science Press. (2019).
Yang, J. et al. China’s conservation program on plant species with extremely small populations (PSESP): progress and perspectives. Biological Conservation 244, 108535, https://doi.org/10.1016/j.biocon.2020.108535 (2020).
Liu, Y. et al. Conservation and threatened status of plant species with extremely small populations in the karst region of southeastern Yunnan, China. Frontiers in Plant Science 15, 1520363, https://doi.org/10.3389/fpls.2024.1520363 (2024).
Genomes, T. P. P. Phylogenetic relationships for flowering plants with genomes sequenced and published. https://www.plabipd.de/plant_genomes_pa.ep (2024).
Marks, R. A., Hotaling, S., Frandsen, P. B. & VanBuren, R. Representation and participation across 20 years of plant genome sequencing. Nature plants 7, 1571–1578, https://doi.org/10.1038/s41477-021-01031-8 (2021).
Zanini, S. F. et al. Pangenomics in crop improvement—from coding structural variations to finding regulatory variants with pangenome graphs. The Plant Genome 15, e20177, https://doi.org/10.1002/tpg2.20177 (2022).
Song, Y., Huang, J.-P., Wang, Y.-J. & Huang, S.-X. Chromosome level genome assembly of endangered medicinal plant Anisodus tanguticus. Scientific Data 11, 161, https://doi.org/10.1038/s41597-024-03007-7 (2024).
Wang, B., Zhang, R., Sun, W. & Yang, J. A nearly telomere-to-telomere diploid genome assembly of Firmiana kwangsiensis, a threatened species in China. Scientific Data 11, 1394, https://doi.org/10.1038/s41597-024-04250-8 (2024).
Zhu, Y. et al. A high-quality chromosome-level genome assembly of the traditional Chinese medicinal herb Zanthoxylum nitidum. Scientific Data 11, 1–13, https://doi.org/10.1038/s41597-024-04174-3 (2024).
Wang, X.-F. et al. A telomere-to-telomere genome assembly of Camellia nitidissima. Scientific Data 12, 1–8, https://doi.org/10.1038/s41597-025-05157-8 (2025).
Ma, H. et al. Chromosome‐level genome assembly and population genetic analysis of a critically endangered rhododendron provide insights into its conservation. The Plant Journal 107, 1533–1545, https://doi.org/10.1111/tpj.15399 (2021).
Cai, L. et al. The chromosome-scale genome of Magnolia sinica (Magnoliaceae) provides insights into the conservation of plant species with extremely small populations (PSESP). GigaScience 13, giad110, https://doi.org/10.1093/gigascience/giad110 (2024).
Lan, L. et al. The haplotype-resolved telomere-to-telomere carnation (Dianthus caryophyllus) genome reveals the correlation between genome architecture and gene expression. Horticulture research 11, uhad244, https://doi.org/10.1093/hr/uhad244 (2024).
Belton, J.-M. et al. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98, https://doi.org/10.17632/c6bg4cbggn.1 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Jin, J.-J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome biology 21, 1–31, https://doi.org/10.1186/s13059-020-02154-5 (2020).
Pryszcz, L. P. & Gabaldón, T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic acids research 44, e113–e113, https://doi.org/10.1093/nar/gkw294 (2016).
Chen, J.-D. et al. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Horticulture Research 7 https://doi.org/10.1038/s41438-020-0288-2 (2020).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome biology 20, 1–18, https://doi.org/10.1186/s13059-019-1905-y (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic acids research 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 1–9, https://doi.org/10.1186/1471-2105-5-59 (2004).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome research 18, 188–196, http://www.genome.org/cgi/doi/10.1101/gr.6743907 (2008).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC bioinformatics 6, 1–11, https://doi.org/10.1186/1471-2105-6-31 (2005).
Cook, D. E. et al. Long-read annotation: automated eukaryotic genome annotation based on long-read cDNA sequencing. Plant physiology 179, 38–54, https://doi.org/10.1104/pp.18.00848 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Zhang, R.-G. et al. TEsorter: an accurate and fast method to classify LTR-retrotransposons in plant genomes. Horticulture Research 9, uhac017, https://doi.org/10.1093/hr/uhac017 (2022).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Molecular biology and evolution 34, 2115–2122, https://doi.org/10.1093/molbev/msx148 (2017).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics, proteomics & bioinformatics 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Database resources of the national genomics data center, China national center for bioinformation in 2022. Nucleic Acids Research 50, D27–D38 https://doi.org/10.1093/nar/gkab951 (2022).
Genome Sequence Archive (GSA). Camellia fascicularis genome sequencing. https://ngdc.cncb.ac.cn/gsa/browse/CRA024719 (2025).
Cai, L. Camellia fascicularis Genome. Genome Warehouse (GWH). https://download.cncb.ac.cn/gwh/Plants/Camellia_fascicularis_jinhuacha_GWHFSQE00000000.1 (2025).
NCBI GenBank. https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_051225895.1 (2025).
NCBI GenBank https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_051225905.1 (2025).
Acknowledgements
This work was supported by National Natural Science Foundation of China (32260416), Yunnan Province Xingdian Talent Project “Youth Talent Special Project” Support Project (YNQR-QNRC-2-19-104), and Yunnan Agricultural Basic Research Joint Special General Project (202101BD070001-095). Funders have no role in the design and publication of the manuscript.
Author information
Authors and Affiliations
Contributions
The authors confirm contribution to the paper as follows: study conception and design: H.X., L.C., F.G. Data collection and analysis: H.X., X.W., F.Y., Z.Y., G.Z., P.Y., L.C., F.G. Draft manuscript preparation: H.X., J.K., L.C., F.G. All authors reviewed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, HX., Wu, XQ., Yan, FJ. et al. High quality chromosome level genome assembly of Camellia fascicularis, an endangered plant in China. Sci Data 12, 1573 (2025). https://doi.org/10.1038/s41597-025-05777-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05777-0
