
Abstract
Sarcandra glabra (Thunb.) Nakai, a member of the family Chloranthaceae, has a rich history of use in traditional Chinese medicine. To date, >400 compounds have been isolated and characterized from this plant. Studying the genome of S. glabra is particularly important for understanding the synthesis and functions of these compounds. This study presents the first genome assembly of S. glabra using nanopore, short-read, and high-throughput chromosome conformation capture sequencing. The final genome assembly was 4.78 Gb, and was grouped into 15 chromosomes. The contig and scaffold N50 values were 602 kb and 226.1 Mb, respectively. Genome annotation revealed 41,423 protein-coding genes, with 80.21% (33,223 genes) annotated into different databases and clustered into 15,026 gene families in S. glabra. Phylogenetic analysis indicated that S. glabra formed a distinct clade with C. sessilifolius, C. salicifolius, and C. demersum. The highly contiguous genome assembly of S. glabra provides a valuable genomic resource for elucidating the genetic basis of this plant and for improving the yield and quality of medicinal plants.
Similar content being viewed by others


Background & Summary
Sarcandra glabra (Thunb.) Nakai (Chloranthaceae; Fig. 1) is a traditional Chinese medicinal herb with high medicinal and edible value. It is distributed mainly south of the Yangtze River and grows in damp areas under mountain valleys and forests at an altitude of 420–1500 m. This species is widely used for its immunomodulatory1, anti-inflammatory2, antitumor3, and fracture healing properties owing to its chemical components, which include flavonoids, terpenes, coumarins, phenolic acids, and volatile oils4,5.

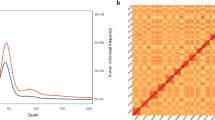
Genome assembly of Sarcandra glabra. (A) S. glabra; (B) Genome-wide Hi-C heatmap of chromatin interaction counts in 100 kb bins. Only sequences anchored on chromosomes are shown. The abbreviations, CSHB01–15, represent the nine chromosomes, and the color bar represents the log2 value of interaction counts.
The family Chloranthaceae, comprising five genera and approximately 70 species, is primarily distributed in tropical and subtropical regions and is used for medicinal purposes and for the extraction of aromatic oils. Chloranthaceae exhibit a degree of primitiveness in their evolutionary relationships. Some genera within this family possess leaf veins containing both tracheids and vessels, reflecting their unique evolutionary journey. However, only one species currently has a genome published in NCBI (Chloranthus sessilifolius, GCA_021018995.1), indicating that much remains to be explored in terms of genomic research and the utilization of genetic resources of Chloranthaceae plants. Within the S. glabra species, two subspecies are officially accepted, viz. Sarcandra glabra subsp. glabra and Sarcandra glabra subsp. brachystachys.
Previous transcriptomic and metabolomic studies have investigated the tissue-specific distribution of terpenoid biosynthesis, the regulatory mechanism of the differential accumulation of flavonoids in leaves and roots, and the mechanism of accumulation of phenylpropanoid-derived compounds in S. glabra6,7,8,9,10; molecular markers have also been reported11. Although regulatory patterns for these compounds have been deduced, it is still necessary to reveal their molecular mechanisms using whole-genome sequencing.
In the current study, nanopore, short-read, and high-throughput chromosome conformation capture (Hi-C) sequencing was used to construct a highly contiguous assembly of the S. glabra genome. High-quality genome assembly facilitates the elucidation of the molecular mechanisms underlying the biosynthesis of beneficial compounds with medicinal value in S. glabra and provides a reference for the development and utilization of S. glabra.
Methods
Sample preparation and DNA extraction
Fresh leaves of S. glabra were collected from the Guangxi Botanical Garden of Medicinal Plants, China (http://www.gxyyzwy.com, Ying Hu, hying@gxyyzwy.com), with a voucher number of YY00902. The samples were stored at −80 °C. Genomic DNA was extracted from the frozen leaves using CTAB (cetyltrimethylammonium bromide) buffer (incubation at 65 °C for 60 min). Then he extracted DNA was purified through phenol/chloroform/isopentyl (25:24:1) extraction, followed by precipitation with isopropyl alcohol and ethanol. The final DNA was resuspended in Tris-EDTA buffer for sequencing.
Library construction and sequencing
Library size selection was carried out using the BluePippin (Sage Science, Beverly, MA, USA), and 1 μg of the genomic DNA (target insert size of 20 kb) was processed for damage repair, end repair, and purification. Nanopore sequencing libraries were prepared using the SQK-LSK109 Ligation Sequencing Kit (ONT, Oxford, UK), following the manufacturer’s instructions. After quality control, the libraries were subjected to PacBio HiFi sequencing.
Two short-read libraries with insert sizes of 270 bp and 500 bp were constructed from high-quality DNA using fragmentation (Covaris, Woburn, MA, USA), end repair, and adaptor ligation, creating circular DNA molecules for rolling-circle amplification to generate DNA nanoballs (DNBs). For Hi-C library preparation, cells were cross-linked with formaldehyde to preserve DNA-protein and protein-protein interactions, followed by fragmentation, end repair, purification, and adaptor ligation. The short-read and Hi-C libraries were sequenced using the DNBSEQ platform (MGI, Shenzhen, China) in paired-end mode.
Genome assembly and quality evaluation
Short-read sequences were processed using SOAPnuke (v1.6.5; -n 0.01, -q 0.1, -l 20, -Q 2, -M 2, -A 0.5)12 to remove low-quality reads and adapter contamination. The genome size was estimated using k-mer analysis with K-mer Analysis Toolkit v2.4.213, followed by genome assessment and heterozygosity estimation using GenomeScope14.
A draft assembly was generated from the ONT data with Necat (GENOME_SIZE = 4455 Mb) and polished using Racon15,16. Short-read data were used to further refine the assembly with Pilon, and redundancy was reduced using Trimdup17. The HiC-Pro v2.5.0 pipeline aligned Hi-C data with the assembled genome contigs to obtain valid interaction pairs18. Juicer was used for sequence alignment, and 3D-DNA was employed to construct a chromosome-level assembly19,20. The final genome quality was assessed using BUSCO v3 with the “embryophyta_odb10” ortholog set21.
Genome annotation
Repeat elements were identified using RepeatMasker v4.0.7 and RepeatProteinMask v4.0.7 (http://www.repeatmasker.org/cgi-bin/RepeatProteinMaskRequest) with the Repbase v21.12 database22,23. A de novo repeat library was created using RepeatModeler, followed by identification of repetitive sequences with RepeatMasker22,23,24. The two predicted repeat sets were merged to generate nonredundant repeat sequences using TEclass2.1.325,26.
Protein-coding gene annotation was performed using Exonerate v2.2.025, Genewise v2.4.1 (https://www.ebi.ac.uk/Tools/psa/genewise/), and Funannotate v1.8.7 (https://github.com/nextgenusfs/funannotate), followed by integration into a comprehensive gene set using EVM (https://github.com/EVidenceModeler/EVidenceModeler/). Non-coding RNAs, including tRNAs, rRNAs, miRNAs, and snRNAs, were identified with tRNAscan-SE27, BLASTN, and INFERRAL (http://infernal.janelia.org/) of Rfam, respectively28. Gene functional annotation was performed using BLAST v2.2.3129 against various databases, including GO database30, KEGG (Kyoto Encyclopedia of Genes and Genomes)31, translation of European Molecular Biology Laboratory32, InterPro33, SwissProt32, and NR (nonredundant protein sequences)34.
Data Records
This Whole Genome Shotgun project has been deposited at GenBank under the accession ASM4507178v1. The version described in this paper is version GCA_045071785.135. The raw genome sequencing data (PacBio and DNBSEQ short reads) have been deposited to NCBI database under the Sequence Read Archive accession number SRP51833936, and the genome annotation was available at figshare with the accession number https://doi.org/10.6084/m9.figshare.28874543.v237.
Technical Validation
Genome assembly
Oxford Nanopore Technologies (ONT) sequencing technology and Hi-C-assisted genome assembly were used to generate highly contiguous genome assemblies of Sarcandra glabra (Thunb.) Nakai. The ONT read data was 124.95 Gb (~28 × coverage), with a mean long-read length and N50 of 27.30 and 33.75 kb, respectively (Table S1). A total of 268.36 Gb of clean short-read sequencing data (~49 × coverage) were used for subsequent polishing (Table S1).
The total length of the final assembly was 4.78 Gb, with a GC content of 38.90% (Table 1), which was close to the genome size estimated by 17-mer analysis (genome size of 4.46 Gb and heterozygosity of 1.10%). The contig N50 and scaffold N50 were approximately 602 kb and 239.7 Mb, respectively, with maximum contig size and scaffold sizes of 3.4 Mb and 424.4 Mb, respectively (Table 1). Fifteen chromosomes were generated by concatenating contigs with a total length of 3.75 Gb based on the Hi-C reads (Table 1).
The interaction signal strength of the genome-wide Hi-C heatmap around the diagonal was higher than that of the off-diagonal signals, demonstrating the high quality of highly contiguous genome assembly (Fig. 1B). BUSCO evaluation indicated that the final genome contained 89.00% complete genes in the “embryophyta_odb10” ortholog set (Table 1), indicating a high degree of completeness for the genome assembly.
Genome annotation
The identified repetitive sequences (233.11 Mb) constituted 37.62% of the reference genome sequence (Table S2). The most abundant repeat types were long terminal repeat (LTR) retrotransposons (26.47%) and DNA elements (10.09%) (Table S2). Prediction yielded 41,423 protein-coding genes in the genome, with an average mRNA length of 3484.43 bp and an average coding sequence length of 1085.04 bp (Table 2). The average exon number was 4.97, with average exon and intron lengths of 330.45 bp and 1133.84 bp, respectively (Table 2). Gene function annotation revealed that 33,223 genes (80.21%) could be annotated into databases such as GO and KEGG (Table 2 and Fig. 2). Non-coding RNAs included 4,354 ribosomal RNAs (rRNAs), 967 transfer RNAs (tRNAs), 639 small nucleolar RNAs, and 143 miRNAs (Table S3).

The functional annotation of the protein-coding genes of the Sarcandra glabra genome. (A) Venn diagram representing the functional annotation in InterPro, KEGG, SwissProt and NR; (B) GO classification statistics; (C) KEGG pathway classification statistics. Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), nonredundant protein sequences (NR).
Code availability
No specifc code was used in this study. The data analyses used standard bioinformatic tools, with parameters being clearly described in Methods. If specific parameters were not provided for the software, default settings recommended by the developer were utilized.
References
He, R. R. et al. Effects of Sarcandra glabra extract on immune activity in restraint stress mice. China J Chin Materia Medica. 34, 100–103 (2009).
Liu, C. P. et al. Combination effect of three main constituents from Sarcandra glabra inhibits oxidative stress in the mice following acute lung injury: a role of MAPK-NF-κB pathway. Front Pharmacol. 11, 2082 (2021).
Zeng, Y. L. et al. The traditional uses, phytochemistry and pharmacology of Sarcandra glabra (Thunb.) Nakai, a Chinese herb with potential for development: review. Front Pharmacol. 12, 652926 (2021).
Yang, X. R. et al. Sarcaglabrin A, a conjugate of C15 and C10 terpenes from the aerial parts of Sarcandra glabra. Tetrahedron Lett. 61, 151916 (2020).
Chu et al. A comprehensive review on the chemical constituents, sesquiterpenoid biosynthesis and biological activities of Sarcandra glabra. Nat Product Bioprosp. 13, 53 (2023).
Jiang, N. et al. Integrated transcriptome and proteome analyses unravel a series of early defence responses in Sarcandra glabra against Colletotrichum gloeosporioides. Funct Plant Biol. 50(12), 1047–1061 (2023).
Li Q. et al. The total biosynthesis route of rosmarinic acid in Sarcandra glabra based on transcriptome sequencing. Plant Physiol Biochem. 109016 (2024).
Xie, D. et al. Transcriptomic and metabolomic profiling reveals the effect of LED light quality on morphological traits, and phenylpropanoid-derived compounds accumulation in Sarcandra glabra seedlings. BMC Plant Bio. 20(1), 476 (2020).
Wu, D. et al. Transcriptional regulation mechanism of differential accumulation offlavonoids in leaves and roots of Sarcandra glabra based on metabonomicsand transcriptomics. China J Chin Materia Medica. 48(21), 5767–5778 (2023).
Wu, D. et al. Tissue specific distribution of terpenoid biosynthesis in Sarcandra glabra based on transcriptome and metabolome analysis. Chin J Biotech. 40(2), 542–561 (2024).
Xu, Y. et al. Transcriptome Characterization and identification of molecular markers (SNP, SSR, and Indels) in the medicinal plant Sarcandra glabra spp. Biomed Res Int. 2021, 9990910 (2021).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7(1), 1–6 (2018).
Mapleson, D. et al. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576 (2017).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33(14), 2202–2204 (2017).
Chen, Y. et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat Commun. 12(1), 60 (2021).
Vaser, R. et al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27(5), 737–746 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9(11), e112963 (2014).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3(1), 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356(6333), 92–95 (2017).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19), 3210–3212 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinform. Chapter 4, Unit 4.10 (2009).
Bao, W. et al. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11 (2015).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA. 117(17), 9451–9457 (2020).
Abrusán, G. et al. TEclass-a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25(10), 1329–1330 (2009).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5), 955–964 (1997).
Sam, G. J. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, 121–124 (2005).
Altschul, S. F. et al. Basic local alignment search tool. J Mol Biol. 215(3), 403–410 (1990).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet. 25, 25 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31(1), 365–370 (2003).
Apweiler, R. et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29(1), 37–40 (2001).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, 225–229 (2011).
Guangxi Botanical Garden of Medicinal Plants. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_045071785.1 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP518339 (2024).
Hu, Y. Gene annotation of the medicinal plant Sarcandra glabra (Thunb.) Nakai. figshare https://doi.org/10.6084/m9.figshare.28874543.v2 (2025).
Acknowledgements
This work was supported by the Guangxi Innovation-Driven Development Project (GuiKe AA18242040), Guangxi Major Science and Technology Project of China (GuikeAA22096021) and Guangxi Key Laboratory of Medicinal Resources Protection and Genetic Improvement (KL2022ZZ03).
Author information
Authors and Affiliations
Contributions
Y.H., M.T., B.H. and X.G. conceived the project and designed the experiments. Y.P., Z.L., Z.Q. and F.Q. conducted the experiments. Y.H., M.T., Y.L., F.W. and X.W. analyzed the results. Y.H. and M.T. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, Y., Tang, M., Peng, Y. et al. Highly contiguous genome of the medicinal plant Sarcandra glabra (Thunb.) Nakai. Sci Data 12, 1508 (2025). https://doi.org/10.1038/s41597-025-05796-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05796-x
