
Abstract
The post-natal development and maturation of the mammalian heart involve highly intricate processes that remain incompletely understood, particularly concerning the molecular signature and roles of the diverse cell types involved. In this study, we present a comprehensive dataset generated from murine hearts at three key post-natal developmental stages using Spatio-Temporal Enhanced Resolution Omics-Sequencing (Stereo-seq), an advanced spatially resolved transcriptomic technology. This dataset encompasses spatial transcriptomes of approximately 0.186 million individual cells within intact sections of murine hearts at post-natal developmental stages. Our dataset serves as a valuable resource for investigating the mechanisms underlying mammalian heart development and maturation. Through initial analyses, we identified distinct cell types and their spatial distributions, including 93,826 cardiomyocytes within a single heart section. This extensive dataset provides researchers with opportunities for data mining and facilitates diverse analyses, including studies on transcriptional regulation, cell-to-cell communication, and the functional activities of genes and signalling molecules during critical phases of heart development.
Similar content being viewed by others

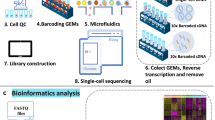

Background & Summary
During the maturation of the mammalian heart, the cardiac chambers undergo a complex series of post-natal developmental processes that remain incompletely understood. As demonstrated by previous studies1,2,3,4,5, these critical processes play a pivotal role in shaping a fully functional heart. Significant changes occur in the size, hemodynamics, and function of cardiovascular structures as the heart transitions from the neonatal to adult stages. Many congenital heart disease conditions arise from genetic defects that disrupt heart development and maturation, leading to alterations in one or more of these processes. Consequently, understanding the molecular signatures and interactions among various cell types during the formation of ventricular chambers from the neonatal stage to adulthood is essential for uncovering the mechanisms underlying these disease conditions.
The rapid development of transcriptomic technologies, including single-cell RNA sequencing (scRNA-seq) and spatial transcriptomic technology, over the past decade has facilitated this type of research at an unprecedented pace by revealing previously uncharacterized cell types involved in cardiogenesis and maturation, focusing on subpopulations of cells isolated using predefined genes6,7,8,9,10. However, each of them has their limitations. ScRNA-seq does not provide spatial information and thus has a disadvantage in studying the complex cellular interplay, intercellular communications and tissue morphogenesis spatially. Recent emerging spatial transcriptomic technologies represent a major advance in high-throughput profiling of in situ gene expression in space, contributing to a systematic understanding of the development and homeostasis of complex tissue molecular architecture11,12. Yet, current spatial transcriptomic methods mostly produce inferior transcriptome sampling to leading scRNA-seq methods and are still unable to achieve single-cell resolution, where the boundaries of spots have no natural correspondence to the boundaries of cells. Therefore, they yield aggregate profiles of small regions encompassing multiple cells and/or portions of cells rather than truly resolving individual cells13, underscoring the need for an approach to achieve single cellular resolution. Generally, image-based spatial transcriptome technologies, such as Merscope, Xenium, and CosMx, offer higher resolution and better RNA detection sensitivity, specificity, and reproducibility than sequencing-based methods. However, these technologies have limited gene profiling capacity: Merscope profiles about 1000 genes, Xenium around 5000 genes, and CosMx approximately 6000 genes. In contrast, sequencing-based technologies provide whole-transcriptome analysis. Although they have lower RNA capture efficiency and detection sensitivity, these can be improved by increasing sequencing depth. As technology advances, the resolution of sequencing-based spatial transcriptome techniques is improving, with examples like 10X Visium (55 μm) and Slide-seq (10 μm)14,15. Recently, a spatio-temporal enhanced resolution omics-sequencing (Stereo-seq) has been developed by scientists at the Beijing Genomics Institute (BGI)16. This technology combines DNA nanoball (DNB) patterned array chips with in situ RNA capture (Fig. 1). It allows high throughput sample transcriptomic profiling of histological sections at an unprecedented nanoscale (with each spot measuring 200 nm in size and spot-spot space distances of 500 or 715 nm), potentially achieving a cellular spatial resolution of 0.22 μm. In summary, Stereo-seq stands out as the spatial transcriptome technique with the highest resolution and widest transcriptome range. Based on Stereo-seq, we further developed a single-cell resolution transcriptomic platform by integrating Stereo-seq with nuclear imputation. The new approach enables us to conduct a systemic analysis of the molecular architectures of murine developing hearts for understanding cardiac maturation.

Workflow of the Stereo-seq procedure. (a) Tissue collection. Hearts of different stages were obtained, frozen in liquid nitrogen, embedded in OCT and made into sections. (b) Stereo-seq chip. Tissue slices were affixed to the Stereo-seq chips made according to the technology developed by BGI. (c) Sequencing and analysis. After cDNA synthesis, sequencing library construction, quality control and analysis, the gene information expressed in the spatial position of the target tissue cells was obtained.
The features of the current dataset
Our dataset is notable for several key features. It encompasses an extensive collection of spatial transcriptomes from 4-chamber heart sections across post-natal cardiac developmental stages, providing detailed information on the sinoatrial node, atrium, ventricle, cardiac conduction system, cardiac trabeculae, and other relevant structures. Utilizing high-resolution Stereo-seq and combining it with nuclear imputation, the dataset incorporates rigorous quality control procedures. Additionally, it leverages an innovative local and global structure-preserving dimensionality reduction technique. This dataset enables the researchers to unravel the intricate differentiation patterns of specific cell types and genes across temporal and spatial dimensions. It also facilitates the exploration of upstream and downstream targets associated with key transcription factors crucial to cardiac development, as well as the molecular characteristics and biological functions of these transcription factors. These insights provide valuable information for future advancements in understanding and addressing cardiac-related disorders.
Methods
Animals
In this study, wild-type C57BL/6J mice at three post-natal developmental stages, including post-natal day 0 (P0), post-natal day 7 (P7), and post-natal day 56 (P56) were used. The mice were supplied by Chengdu Gempharmatech Co. and housed at the Laboratory Animal Center of Southwest Medical University. All procedures involving animal experiments presented in this study complied with ethical regulations for animal research and were approved by the Animal Care and Use Committee of Southwest Medical University, Sichuan (China) (No: 201903183).
Tissue collection for stereo-seq experiment
The hearts of mice at stages P0 (female), P7 (one male and one female), and P56 (two male) were sacrificed, followed by direct dissection. The hearts were cleaned with cooled 1 × PBS buffer and dried with Kimwipes. Subsequently, all heart tissue samples were rapidly frozen in liquid nitrogen and then embedded in Tissue-Tek OCT. Coronal cryosections of the whole heart on four-chamber views, with a thickness of 10 μm, were prepared using a Leica CM1950 cryostat, ensuring that each section contains the sinoatrial node tissue as much as possible. Finally, these tissue sections were affixed to Stereo-seq chips for subsequent experimental procedures. The three slices at the P0 and P7 stages are from the same batch, while those from P56 stage are from another batch.
Generation of stereo-seq chip
This study employed the spatial transcriptomics (STOmics) technology developed by the Beijing Genomics Institute (BGI). It combined DNA nanoball (DNB) chips and in situ RNA capture technology to examine spatial gene expression patterns in tissue sections from specific samples16. A comprehensive protocol outlining this methodology is available on the MOSTA website (https://db.cngb.org/stomics/mosta)17. The initial step involved depositing DNBs containing random barcode sequences onto a photolithographic etched and modified chip. Compared to the bead-based approach18, rolling ring amplification generated DNBs labelled with larger spatial barcode pools while maintaining sequence accuracy. The array was then microscopically imaged and incubated with sequencing primers, generating data that included the coordinate identity (CID) of etched DNBs. Subsequently, hybridization between CIDs and oligonucleotides containing polyT sequences at specific locations to establish molecular identity (MID). The CID sequences and their respective coordinates for all DNB were identified using a base calling method following the manufacturer’s guidelines for the MGI DNBSEQ-Tx sequencer. After sequencing, the capture chip was divided into smaller chips (10 mm × 10 mm) for further processing. At this stage, duplicated CIDs corresponding to non-adjacent spots were removed.
Stereo-seq library preparation and sequencing
RNA quality assessment
In order to preserve the integrity of RNA in the sample, it was recommended to conduct an evaluation of RNA quality. This involved collecting 10–20 pieces of 10 μm sections and placing them in a pre-cooled 1.5 ml Eppendorf tube at −20 °C. Subsequently, the RNA should be isolated from the sections. A qualified assessment of RNA integrity was indicated by an RNA integrity number (RIN) value of ≥7.
Tissue fixation, imaging and permeabilization
Tissue samples were affixed to the surface of the Stereo-seq chip and incubated at 37 °C for a period of 3–5 min. Three P0 slices were affixed on one chip, four P7 slices were affixed on two chips, and two P56 slices were affixed on two chips. All slices on a single chip were from the same mouse, while different chips were sourced from different mice. Subsequently, the sections were treated with methanol and incubated at −20 °C for 40 min before starting the initiation of the Stereo-seq library preparation process. In certain cases, the aforementioned sections were stained with a nucleic acid dye (Thermo Fisher, Q10212) and imaged using a Ti-7 Nikon Eclipse microscope before in situ capture at the FITC channel. After imaging, tissue permeabilization was performed with 0.1% pepsin (Sigma, P7000) in 0.01 M HCl buffer (pH = 2) at 37 °C incubator for varying durations of 3, 6, 9, 12, 15, 18, and 21 min. The optimal permeabilization time was 6 min for P0 and P7 heart sections, and 18 min for P56 heart sections.
Reverse transcription and amplification
The spatially designated probe located on the surface of the STOmics chip was able to capture the released mRNA molecules. Subsequently, a reverse transcription process was carried out, resulting in the release of cDNA. The amplified cDNA, which was used for DNB generation, served as a template for library preparation and was sequenced alongside CIDs. Finally, it was sequenced on the MGI DNBSEQ-Tx sequencer with the read length indicated.
Stereo-seq raw data processing
Fastq files were produced using the MGI DNBSEQ-Tx sequencer. The read 1 contained the CID and MID (CID: 1–25 bp, MID:26–35 bp), while the read 2 comprised the cDNA sequences. Quality control procedures were applied to both read 1 and read 2. The CID sequences in read 1 were compared with the barcode sequences on the chip, and read pairs containing valid CIDs were isolated. For read pairs containing valid CIDs, the CID sequence was converted into spatial position information on the slice and incorporated into the read ID of read 2. Subsequently, valid reads in read 2 were filtered out as the final Clean Reads. Finally, this information was used to generate a CID-containing expression profile matrix. The entire process was incorporated into a publicly available pipeline STOmics Analysis Workflow (SAW, v5.1.4) available at https://github.com/BGIResearch/SAW. Then, leverage nucleic acid staining from the same section to segment cells by projecting the staining image onto the Stereo-seq chips. To accomplish this, the total UMI in each DNB spot, which contained a specific spatial coordinate, was summed to generate a spatial density matrix. This matrix was then converted into an image where each pixel corresponded to one DNB and the total UMI of the DNB spot determined the grayscale of the pixel. Then register the DNB image with the nucleic acid staining image manually. After comparison, the Scikit-image package (v0.18.1, https://scikit-image.org) was used for cell segmentation analysis. Ultimately, by analyzing this sequenced data, we achieved a spatially resolved transcriptome with an impressive resolution of 500 nm or 715 nm.
Data Records
The raw data have been deposited into the Spatial Transcript Omics DataBase (STOmics DB, https://db.cngb.org/stomics/project) under accession ID STT000009019. All processed data have been uploaded to the CNGB Sequence Archive FTP public service. These data include sequencing paired fastq files and spatial files. The dataset has also been submitted to the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE29865020. Each heart’s data contains two raw data files in fastq.gz format (read 1 and read 2). In addition, four processed data files are available in the dataset, containing tissue image files in tif format, raw gene expression matrix files with the suffix gem.gz, tissue raw gene expression matrix files with the suffix TissueCut.gem.gz, and cell cluster information with the suffix Seurat.rds. The GEO accession IDs are GSM9019992 for the P0 heart, GSM9019993 for the P7-1 heart, GSM9019994 for the P7-2 heart, GSM9019995 for the P56-1 heart, and GSM9019996 for the P56-2 heart.
Technical Validation
The SAW was utilized for conducting sequencing data quality control, genome alignment, and gene expression quantification. Quality control procedures were applied to Read1 and Read2. Initially, the CID sequences in Read1 were compared with the barcode sequences on the chip and read pairs containing valid CIDs were isolated. Then, these valid CID sequences were transformed into spatial position information on the slice and incorporated into the read ID of Read2. Subsequently, valid reads in Read2 were filtered to obtain the final Clean Reads. The statistical quality control outcomes are presented in Table 1. The Clean Reads were aligned to the reference genome, and the counts of reads mapped to exon regions, intron regions, and intergenic regions were determined. The statistical results are displayed in Table 2. Unique Mapping Reads were assigned to genes, duplicate MIDs were eliminated, and the expression levels of all genes were calculated with MID correction. The statistical results are presented in Table 3.
In STOmics, the Bin serves as the fundamental unit for data statistics, representing a fixed-size region (200 nm size per spot and 500 or 715 nm spot-spot space distance). The Bin sizes of P0 and P7 are 500 nm, and that of P56 is 715 nm. The number of DNBs within each region is accumulated, and the regions do not overlap. The Bin number corresponds to a single DNB number. Each DNB on the STOmics chip is a pixel on the gene expression heatmap, and the analysis unit at this point is Bin1, meaning each Bin contains data from a single DNB. When adjacent N × N DNB data is combined and displayed as a single pixel on the gene expression heatmap, the analysis unit becomes Bin N, and the size is 500/715 nm × N. For example, Bin 100 represents an analysis unit containing data from 100 × 100 = 10,000 DNBs. The choice of Bin size can be adjusted based on cell size and gene number. Statistical results of genes captured with different Bin sizes are presented in Table 4.
Here, the Bin 50 gene expression matrix was taken as an example. Seurat v4.0.0 (https://satijalab.org/seurat) was used to conduct matrix statistics, cell clustering, marker gene screening, and other analyses on gene expression data. As shown in Fig. 2, gene expression quantity (nCount) and gene number (nFeature) in the gene expression matrix are tallied and presented in the spatial position of the sample. Principal-component analysis (PCA)21 was utilized to reduce the data’s dimensionality, followed by applying the UMAP algorithm for cell clustering. The differentially expressed genes of each cluster of cells with others were calculated separately by Seurat software, and the top 10 differential genes were screened as the marker genes. Figure 3 shows the top 3 marker gene heatmaps that define cardiomyocytes, fibroblasts, endothelial cells, epithelial cells, macrophages, granulocytes, neurons, and T cells. Figure 4 shows the top 3 marker gene violin diagrams, which mark gene expression levels with specific numbers. Numbers represent different clusters, and the specific corresponding classification is in the uploaded data. Figure 5 shows the spatial arrangement of cell clusters on sample tissues to which the top 1 gene belonged. After obtaining different cell groups by clustering analysis based on Seurat, SingleR (v1.6.1, https://bioconductor.org/packages/3.18/bioc/html/SingleR.html) can compare each cluster with public databases and score the cluster according to the similarity of gene expression between cluster22, with the MouseRNAseqData, ImmGenData, and CellMaker2.0 as the reference genomes. Bins can be annotated into different cell groups by scoring matrix results. The spatial location of each cluster was visualized by comparing it with the tissue image and manually annotating it, then the findings are presented in Fig. 6. As shown in Fig. 6, each dot represents a cell and displays the location of sample sections.

Gene expression quantity (nCount) and gene number (nFeature) statistics of P0, P7 and P56 stages (Bin 50). (a) nCount and nFeature at P0 stage. (b,c) nCount and nFeature at P7 stage. (d,e) nCount and nFeature at P56 stage.

Heatmaps of top 3 marker gene at P0, P7 and P56 stages (Bin 50). (a) P0 stage. (b,c) P7 stage. (d,e) P56 stage.

Violin diagram of top 3 marker gene at P0, P7 and P56 stages. (a) P0 stage. (b,c) P7 stage. (d,e) P56 stage.

The top 1 marker gene of each cellular cluster is depicted within the respective cluster location on the sample sections (Bin 50). (a) P0 stage. (b,c) P7 stage. (d,e) P56 stage. The red dots indicate the spatial location of the cell cluster associated with the marker gene within the tissue sample section.

Definition of cell types. Cell clustering results of P0 (a), P7 (b, c) and P56 (d,e) stages (Bin50). The figure on the left shows the UMAP algorithm cell clustering results; On the right is the arrangement of cell groups in the sample tissue section space.
At the P0 stage, of all the 25458 cells, there were 15452 cardiomyocytes (60.70%), 3938 fibroblasts (15.5%), 2285 endothelial cells (8.98%), 1861 epithelial cells (7.31%), 390 granulocytes (1.53%), and 1532 neurons (6.02%). When it came to P7 stage, there were 75935 cells totally, including 37768 cardiomyocytes (49.74%), 1660 adipocytes (2.19%), 6372 erythrocytes (8.39%), 25378 fibroblasts (33.42%), 959 granulocytes (1.26%), 3590 neurons (4.73%), and 208 macrophages (0.27%). At P56 stage, among the 84654 cells, it existed 40606 cardiomyocytes (47.97%), 2873 adipocytes (3.39%), 6823 endothelial cells (8.06%), 17740 fibroblasts (20.96%), 657 granulocytes (0.78%), 466 macrophages (0.55%), 14293 neurons (16.88%), 255 T cells (0.30%), and 941 erythrocytes (1.11%).
Code availability
All Stereo-seq analyses were performed using STOmics Analysis Workflow (SAW, v5.1.4, https://github.com/BGIResearch/SAW), Scikit-image package (v0.18.1, https://scikit-image.org), Seurat (https://satijalab.org/seurat), and SingleR (v1.6.1, https://bioconductor.org/packages/3.18/bioc/html/SingleR.html).
References
Taber, L. A. Mechanical aspects of cardiac development. Prog Biophys Mol Biol 69, 237–255, https://doi.org/10.1016/s0079-6107(98)00010-8 (1998).
Sedmera, D., Pexieder, T., Vuillemin, M., Thompson, R. P. & Anderson, R. H. Developmental patterning of the myocardium. Anat Rec 258, 319–337, 10.1002/(sici)1097-0185(20000401)258:4<319::Aid-ar1>3.0.Co;2-o (2000).
Bartman, T. & Hove, J. Mechanics and function in heart morphogenesis. Dev Dyn 233, 373–381, https://doi.org/10.1002/dvdy.20367 (2005).
Moorman, A. F., Christoffels, V. M., Anderson, R. H. & van den Hoff, M. J. The heart-forming fields: one or multiple? Philos Trans R Soc Lond B Biol Sci 362, 1257–1265, https://doi.org/10.1098/rstb.2007.2113 (2007).
Zhang, W., Chen, H., Qu, X., Chang, C. P. & Shou, W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C Semin Med Genet 163c, 144–156, https://doi.org/10.1002/ajmg.c.31369 (2013).
Li, G. et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Developmental Cell 39, 491–507, https://doi.org/10.1016/j.devcel.2016.10.014 (2016).
Lescroart, F. et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 359, 1177–1181, https://doi.org/10.1126/science.aao4174 (2018).
Goodyer, W. R. & Wu, S. M. Fates Aligned: Origins and Mechanisms of Ventricular Conduction System and Ventricular Wall Development. Pediatr Cardiol 39, 1090–1098, https://doi.org/10.1007/s00246-018-1869-9 (2018).
Xiong, H. et al. Single-Cell Transcriptomics Reveals Chemotaxis-Mediated Intraorgan Crosstalk During Cardiogenesis. Circulation Research 125, 398–410, https://doi.org/10.1161/CIRCRESAHA.119.315243 (2019).
Tyser, R. C. V. et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science 371, eabb2986, https://doi.org/10.1126/science.abb2986 (2021).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82, https://doi.org/10.1126/science.aaf2403 (2016).
Eng, C. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239, https://doi.org/10.1038/s41586-019-1049-y (2019).
Longo, S. K., Guo, M. G., Ji, A. L. & Khavari, P. A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nature Reviews Genetics https://doi.org/10.1038/s41576-021-00370-8 (2021).
Bressan, D., Battistoni, G. & Hannon, G. J. The dawn of spatial omics. Science 381, eabq4964, https://doi.org/10.1126/science.abq4964 (2023).
Lim, H. J., Wang, Y., Buzdin, A. & Li, X. A practical guide for choosing an optimal spatial transcriptomics technology from seven major commercially available options. BMC Genomics 26, 47, https://doi.org/10.1186/s12864-025-11235-3 (2025).
Chen, A. et al. Large field of view-spatially resolved transcriptomics at nanoscale resolution. bioRxiv, 2021.2001.2017.427004, https://doi.org/10.1101/2021.01.17.427004 (2021).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792.e1721, https://doi.org/10.1016/j.cell.2022.04.003 (2022).
Burgess, D. J. Technology: Bead capture for single-cell transcriptomics. Nat Rev Genet 16, 195, https://doi.org/10.1038/nrg3919 (2015).
Zheng, Y., Ou, X., Sun, T., Lei, M. & Tan, X. (2024).
NCBI GEO https://identifiers.org/geo/GSE298650 (2025).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296, https://doi.org/10.1186/s13059-019-1874-1 (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287, https://doi.org/10.1089/omi.2011.0118 (2012).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81700308 to X.O., 31871181 to M.L., 82470323 to X.T., and 82270334 to X.T.), Department of Science and Technology of Sichuan Province of China (2024YFFK0180 to X.O., 2024JDHJ0051 to X.T., 2025ZNSFSC0052 to X.T., 2025HJPJ0005 to M.L.), and Luzhou Science and Technology Program (2023JYJ004 to T. C., 2024LZXNYDJ006 to X.T., 2023RCX174 to X.O.).
Author information
Authors and Affiliations
Contributions
Y.Z., X.O., T.S., Z.P., Y.Y., M.L., X.T., T.L., and T.C. carried out the experiments. Y.Z., X.O., T.S., Z.P., and T.C. carried out the data process and data analysis. Y.Y., M.L., X.T., T.L., and T.C. designed the experiments. Y.Z., M.L., T. S., and T.C. drafted the manuscript. M.L., X.O., Y.Y., X.T., and T.C. revised and edited the manuscript. All authors have made a substantial contribution to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zheng, Y., Sun, T., Pu, Z. et al. A dataset of spatially resolved transcriptomics of post-natal cardiac development in mice. Sci Data 12, 1531 (2025). https://doi.org/10.1038/s41597-025-05838-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05838-4
