
Abstract
Chelidonium majus is a herbaceous plant of significant medicinal value, which has been widely distributed in Europe, Asia, and Northern Africa. However, its genome remains uncharacterized. Herein, we present a high-quality chromosome-scale genome for C. majus with a size of 1.06 Gb, and 91.21% of the sequences anchored onto 6 chromosomes, comprising 1,520 contigs with an N50 of 106.65 Mb. The genome is predicted to contain 25,203 protein-coding genes, with 98.2% have been functional annotated. The completeness of the genome is highlighted by a BUSCO score of 97.6%. This high-quality genome assembly provides a vital resource for future gene screening, drug discovery, and pharmacological exploration in C. majus.
Similar content being viewed by others

Background & Summary
Chelidonium majus L. (Papaveraceae), commonly known as celandine, greater celandine, celandine poppy, rock poppy, felonwort, and swallow-wort, is a short-lived hemicryptophyte and can reach up to 1 m in height with a branched, sparsely hairy stem1. It prefers moist, nitrogen-rich soils and grows in lowlands, foothills, gardens, and roadsides, which is widely distributed in Europe, Asia, and Northern Africa2,3. Researches have shown that it has pharmacologically significant functions in both Western phytotherapy and traditional Chinese medicine4,5. In Chinese herbal medicine, it is employed to address whooping cough, blood stasis, chronic bronchitis, asthma, jaundice, gallstones, and gallbladder discomfort, as well as to stimulate diuresis in cases of edema and ascites1,4.
In addition to its use in human medicine, C. majus also has the potential to treat parasitic diseases in aquatic animals. For example, in vivo assays showed that the three ethanolic extract of C. majus named chelidonine, chelerythrine and sanguinarine, could be 100% effective for the elimination of Trichodina at the concentrations of 1.0, 0.8, and 0.7 mg/L, respectively6. C. majus also can lead to the death of Ichthyophthirius multifiliis theronts in vitro7. The ethanol extract from C. majus whole plant also has shown the significant anthelmintic activity against Dactylogyrus intermedius8.Meanwhile, different parts of C. majus exhibit varying antioxidant capacity and cytotoxic effects. In the ABTS antioxidant assay, the flower extract showed the highest efficacy of 57.94%, while the leaf, pod, and root extracts displayed activities of 39.10%, 36.08%, and 28.88% respectively. However, the highest cytotoxic effect also was observed in the flower extracts9. The major pharmacologically relevant components of C. majus include isoquinoline alkaloids–berberine, chelidonine, chelerythrine, coptisine, and sanguinarine10.
This research first presents a high-quality, chromosome-level assembly for C. majus, generated by a combined approach utilizing PacBio high-fidelity (HiFi) sequencing and high-throughput chromosome conformation capture (Hi-C) technology. In total, we generated 68.14 Gb of Illumina paired-end short reads, 37.40 Gb of PacBio HiFi reads, and 114.28 Gb of Hi-C reads (Table 1). The 17-mers were counted as 50,337,123,571 from the Illumina short reads, and the k-mer depth was 45 (Table 2). The assembled genome assisted by Hi-C amounted to 1.06 Gb, comprising 1,520 contigs, with an N50 of 106.65 Mb (Table 3). 69.27% of the assembled genome comprised repeat sequences (Table 4). A total of 25,203 protein-coding genes were identified and 98.2% of them were successfully predicted (Tables 5 and 6). Additionally, the genome completeness was evaluated by BUSCO scoring, which showed a remarkable level of completeness of 97.6% (Table 8). With the publish of this high-quality reference genome, it can facilitate the discovery of novel pharmaceuticals by identifying genes responsible for bioactive alkaloid synthesis. Meanwhile, it can advance biomedical research by elucidating the biosynthetic pathways and regulatory mechanisms of its active compounds, thereby enhancing our understanding of the relationship between genome and metabolic pathways.
Methods
Sample collection
All specimens were collected following the guidelines of the Earth Biogenome Project (https://www.earthbiogenome.org/sample-collection-processing-standards-2024). Fresh leaves and roots of Chelidonium majus were collected from fields (30.86°N, 120.19°E) in Huzhou, Zhejiang, China in March 2024. Samples were immediately stored at −80°C until DNA extraction. Each sample was associated with a properly preserved voucher specimen, deposited in Zhejiang Institute of Freshwater Fisheries under catalog number (ZIFF-CM-001 and ZIFF-CM-002).
DNA/RNA extraction
The leaves samples were used for DNA isolation by standard CTAB method. First, samples were lysed in 1000 μL of CTAB buffer and supplemented with 20 μL lysozyme, followed by incubation at 65 °C for 2-3 hours with periodic mixing. After centrifugation, 950 μL of supernatant was extracted with an equal volume of phenol: chloroform: isoamyl alcohol (25:24:1), followed by a second extraction using chloroform: isoamyl alcohol (24:1). The DNA was then precipitated by adding 3/4 volume isopropanol and incubating at −20 °C. Subsequent steps included centrifugation, washing the pellet twice with 75% ethanol, and air-drying the DNA under sterile conditions. The purified DNA was resuspended in 51 μL ddH2O, with optional heating at 55–60 °C to facilitate dissolution. Finally, residual RNA was removed by adding 1 μL RNase A and incubating at 37 °C for 15 minutes. Both leaves and roots were subjected to RNA isolation using Trizol reagent (Invitrogen, CA, USA). The quantity of DNA and RNA were examined by a Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, USA) and a Bioanalyzer 2100 system (Agilent Technologies, CA, USA), respectively. The results showed that the concentration of DNA was 232 ng/μL, with the A260/A280 and A260/A230 values of 1.80 and 2.10, respectively. The concentration of RNA was 160 ng/μL, with the RIN value of 6.9. The quality of extracted DNA and RNA were evaluated using agarose gel electrophoresis and NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, USA). DNA and RNA concentrations were determined to be 253.22 ng/μL and 168.40 ng/μL, respectively.
Library preparing and sequencing
For the short reads sequencing, the qualified DNA sample was randomly fragmented using the Covaris ultrasonic disruptor, followed by library generation with an insert size of 350 bp. For Hi-C sequencing, Hi-C libraries were prepared and constructed according to the previously described methods11. After quality inspection, all the constructed libraries were subjected to 150 bp paired-end (PE) sequencing on the Illumina NovaSeq 6000 platform (Illumina, CA, USA). For PacBio sequencing, a SMRTbell library was constructed using SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, CA, USA). AMPure PB Beads were used to concentrate and purify the library. The constructed library was then sequenced on the PacBio Sequel II platform. For transcriptome sequencing, the TruSeqTM RNA Sample Preparation Kit (Illumina, CA, USA) was used to construct RNA-seq transcriptome libraries and followed by sequencing on the Illumina NovaSeq 6000 platform. Besides, Iso-Seq Express 2.0 Kit (Pacific Biosciences, CA, USA) and Kinnex full-length RNA Kit (Pacific Biosciences, CA, USA) were used to synthesis cDNA and construct library, respectively. The library was then subjected to sequencing with the PacBio Sequel II platform. In summary, 68.14 Gb short reads, 37.40 Gb PacBio reads, 114.28 Hi-C reads, and 47.11 RNA-seq reads of Chelidonium majus were generated in this study (Table 1).
Genome size and heterozygosity estimation
Adaptors and low-quality reads were removed from the raw data using fastp (v0.21.0)12. The clean data was employed for genome size estimation. K-mer analysis was conducted using the software Jellyfish (v2.2.7)13. K-mer 17 was used to conduct survey analysis. The results showed that the genome size of C. majus was estimated to be 1,118.6 Mb, with the heterozygous ratio of 1.07% (Table 2).
De novo Genome assembly and chromosome construction
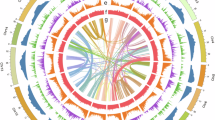
For the de novo genome assembly, a hybrid strategy was adopted, combining the both clean PacBio HiFi reads and Illumina Hi-C reads. First, use the CCS (https://github.com/PacificBiosciences/ccs, parameters: min-rq = 0.99) to perform quality control on the 37.4 Gb raw HiFi sequencing data. The resulting high-fidelity reads were subsequently assembled into contigs using the Hifiasm (v0.19.8)14 with default parameters. To achieve chromosome-level scaffolding, the contig assembly was integrated with the sequenced 114.28 Gb Hi-C data through the ALLHiC pipeline15, including five steps: pruning, partition, rescue, optimization, building. Final manual refinement was performed using Juicebox (v1.11.08)16. The heatmap of both intra- and inter-chromosomal interactions was visualized (Fig. 1). A 918,794,832 bp (91.21%) of sequences were successfully anchored onto 6 pseudo-chromosomes. Estimated genome information in the C-value database at Kew (https://cvalues.science.kew.org/search) showed that the estimated genome size of 1.107 Gb and chromosome number of 2n = 2x = 12, which provided independent support for the assembly in this study. Finally, the assembled genome amounted to 1.06 Gb, comprising 1,520 contigs, with an N50 of 106.65 Mb (Table 3). The circos plot of C. majus genome was shown in Fig. 2.

Hi-C interaction analysis.

Circos plot of Chelidonium majus genome illustrating from outside to inside: (a) chromosome length, (b) gene density, (c) TE density, (d) GC content, (e) collinearity.
Repetitive sequence annotation
Repetitive sequence annotation was performed using a combination of homology-based sequence alignment and de novo prediction approaches. For the homology-based sequence alignment, RepeatMasker (v4.1.6)17 was employed to search against the Repbase TE library18 to identify sequences similar to known repetitive elements. For the de novo prediction, a de novo repetitive sequence library was first constructed using RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html), followed by de novo repeat prediction. Finally, a total of 697,778,264 bp of repetitive sequences were identified in the assemble genome of C. majus (Table 4), including short interspersed nuclear element (SINE, 1.07%), short interspersed nuclear element (LINE, 5.92%), long terminal repeat (LTR, 45.08%), DNA transposon (15.79%), and unknown element (1.00%), which occupied 69.27% of the genome.
Gene structure prediction
For the gene structure prediction, a comprehensive approach combining de novo, homology-based, and transcriptome-based methods was used to predict genes within the assembled genome. For homology-based prediction, protein sequences from Arabidopsis thaliana (Atha) (Col-PEK1.5), Macleaya cordat (Mcor) (GCA 002174775.1), and Papaver somniferum (Psom) (GCF 003573695.1) were collected for mapping onto the C. majus genome using TBLASTN19 with an e-value ≤ 10−5. For the de novo prediction, Augustus (v3.5.0)20 and SNAP (http://homepage.mac.com/iankorf) were used to predict gene coding regions with default parameters. For transcriptome-based gene prediction, Trinity(v2.8)21 was first used to perform transcriptome assembly, followed by predicting the gene structure by PASA(v2.5.2)22. EVidenceModeler(EVM)v1.1.1(http://evidencemodeler.sourceforge.net) was employed to merge the gene sets predicted by the various methods into a non-redundant and more comprehensive gene set. Subsequently, the PASA pipeline (http://pasa.sourceforge.net)23 was employed to refine the EVM annotations by incorporating transcriptome assembly data to produce the final gene set. A total of 25,203 protein-coding genes were identified. The average CDS length was 1,258.59 bp. The average exon number per gene was 5.11 with an average exon length of 246.34 bp and average intron length of 596.13 bp (Table 5). AGAT Tool kit (https://github.com/NBISweden/AGAT) also was used to assess this genome. The result showed that the number of genes containing only 3’UTR is 808, the number of genes containing only 5’UTR is 238, and the number of genes containing both 3’UTR and 5’UTR is 14,369. The number of single exon genes was 4766.
Gene function prediction
For the gene function prediction, the protein sequences were aligned against known protein libraries including National Center for Biotechnology Information (NCBI) Non-Redundant (NR), Swiss-Prot24, InterPro25, and Pfam26 databases using BLAST19 with an e-value ≤ 10−5 (access time: July 10, 2024). Blast2GO(v6.0)27 was employed to annotate functions and pathways based on the Gene ontology (GO)28 and Kyoto Encyclopedia of Genes and Genomes (KEGG)29 databases (access time: July 10, 2024). A total of 24,749 protein-coding genes were successfully predicted (Table 6 and Fig. 3).

Venn diagram of function annotations from different databases.
Non-coding RNA annotation
For the non-coding RNA annotation, tRNAscan-SE30 was used for the tRNA prediction and ribosomal RNAs (rRNAs) were identified by BLAST. miRNA and snRNA were predicted by using Infernal (v1.1)31 against the Rfam database32. The results of non-coding RNA annotation were shown in Table 7.
Data Records
The reads generated in this study have been deposited in the Sequence Read Archive (SRA) under BioProject accession PRJNA1155221(DNA sequence of Illumina pair-end short reads: SRR3050527733, Hi-C reads: SRR3050527834, SRR3050527935, SRR3050528036, PacBio HiFi reads: SRR3050527237, and RNA-Seq reads: SRR3050527338, SRR3050527439, SRR3050527540, SRR3050527641). The genome assembly have been deposited in the GenBank database under the accession number JBGVUA00000000042. The annotation result files have been deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.28407596)43.
Technical Validation
Various different methods were used to ascertain the completeness and accuracy of the Chelidonium majus genome. First, The Hi-C heatmap validated the accuracy of the genome assembly by displaying distinct signals for the 6 pseudo-chromosomes, which indicated their relative independence from one another (Fig. 1). Second, the benchmarking universal single-copy orthologues (BUSCO) v5.4.5 analysis with the “embryophyta_odb10” data set further validated the completeness and accuracy of the assembled genome and annotated genes, achieving a score of 97.6% and 95%, which demonstrates robust annotation quality (Table 8). Third, Illumina paired-end short reads were aligned to the assembled genome using bwa44. Results showed that the read mapping rate was 98.73% and genome coverage was 99.98%, indicating high consistency between reads and assembled genomes (Table 9).
Finally, the QV (quality value) of the assembled genome calculated by Merqury45 was 46.7778, suggesting the genome-wide error rate was only 0.0021% (Table 10). All these results suggested this C. majus assembled genome was of high quality.
Code availability
No custom code was used for this study. All data analyses were performed using published bioinformatics software, which were thoroughly described in the Methods section.
References
Ciric, A., Vinterhalter, B., Šavikin, K., Soković, M. & Vinterhalter, D. Chemical analysis and antimicrobial activity of methanol extracts of celandine (Chelidonium majus L.) plants growing in nature and cultured in vitro. Arch. Biol. Sci. 60 (2008).
Korzeniak, U. et al. Ecological indicator values of vascular plants of Poland. Kraków. W. Szafer Institute of Botany, Polish Academy of Science, 183 (2002).
Monavari, S. H., Shahrabadi, M. S., Keyvani, H. & Bokharaei-Salim, F. Evaluation of in vitro antiviral activity of Chelidonium majus L. against herpes simplex virus type-1. Afr. J. Microbiol. Res. 6, 4360–4364 (2012).
Gilca, M., Gaman, L., Panait, E., Stoian, I. & Atanasiu, V. Chelidonium majus–an integrative review: traditional knowledge versus modern findings. Complement Med Res 17, 241–248 (2010).
Maji, A. K. & Pratim Banerji, P. B. Chelidonium majus L.(greater celandine)-a review on its phytochemical and therapeutic perspectives. J. Herb. Med. (2015).
Yao, J. Y. et al. Isolation of bioactive components from Chelidonium majus L. with activity against Trichodina sp. Aquaculture 318, 235–238 (2011).
Alijanpour, Z. et al. In vitro study of effects of alcoholic extract of Chelidonium majus L. on Ichthyophthirius multifiliis theronts. Journal of Fisheries 75, 405–417 (2022).
Yao, J. Y. et al. In vivo anthelmintic activity of chelidonine from Chelidonium majus L. against Dactylogyrus intermedius in Carassius auratus. Parasitol. Res. 109, 1465–1469 (2011).
Nile, S. H. et al. Comparative analysis of metabolic variations, antioxidant potential and cytotoxic effects in different parts of Chelidonium majus L. Food Chem. Toxicol. 156, 112483 (2021).
Zielińska, S. et al. Greater celandine’s ups and Downs− 21 centuries of medicinal uses of Chelidonium majus from the viewpoint of today’s Pharmacology. Front. Pharmacol. 9, 299 (2018).
Belton, J.-M. et al. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Nishimura, D. RepeatMasker. Biotech Software & Internet Report 1, 36–39 (2000).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 1–6 (2015).
Mount, D. W. Using the basic local alignment search tool (BLAST). Cold spring harbor Protocols 2007, pdb-top17 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Gene Ontology, C. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 (2003).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505277 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505278 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505279 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505280 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505272 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505273 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505274 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505275 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30505276 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_048932765.1 (2025).
Bu, X. Chromosome-level genome assembly of Chelidonium majus. Figshare https://doi.org/10.6084/m9.figshare.28407596 (2025).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 https://doi.org/10.6084/M9.FIGSHARE.963153.V1 (2013).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 1–27 (2020).
Acknowledgements
This work was supported by grants from Hubei Provincial Key Laboratory of Fish. Resources Protection in the Three Gorges Project (2021045-ZHX), Zhejiang Technology Collaboration Project of “Jian Bing Ling Yan” (2024C02005), Huzhou Key Research and Development Project (2023ZD2032), Huzhou Municipal Public Welfare Agricultural Applied Research Project (2022GZ31), and Exploratory Project of Zhejiang Institute of Freshwater Fisheries (2024TSX02).
Author information
Authors and Affiliations
Contributions
J.Y., A.Z. and H.Q. designed the experiments. X.P. and L.H. made the experiments. J.C. and C.N. analyzed the experimental data. Y.Z. and J.Z. helped with the data analysis. X.B. wrote the paper. W.S., X.H. and C.K. reviewed and revised the paper. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bu, X., Peng, X., Chen, J. et al. A chromosome-level genome assembly of the Chinese herbal medicine Chelidonium majus. Sci Data 12, 1642 (2025). https://doi.org/10.1038/s41597-025-05928-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05928-3
