
Abstract
In order to assess the impact of the Fukushima derived Pu isotopes on seawater, a new analytical method to rapidly determine Pu isotopes in seawater by SF-ICP-MS including Fe(OH)2 primary co-precipitation, CaF2/LaF3 secondary co-precipitation and TEVA+UTEVA+DGA extraction chromatographic separation was established. High concentration efficiency (~100%) and high U decontamination factor (~107) were achieved. The plutonium chemical recoveries were 74–88% with the mean of 83 ± 5%. The precisions for both 240Pu/239Pu atom ratios and 239+240Pu activity concentrations were less than 5% when 15 L of seawater samples with the typical 239+240Pu activity of the Northwest Pacific were measured. It just needs 12 hours to determine plutonium using this new method. The limit of detection (LOD) for 239Pu and 240Pu were both 0.08 fg/mL, corresponding to 0.01 mBq/m3 for 239Pu and 0.05 mBq/m3 for 240Pu when a 15 L volume of seawater was measured. This method was applied to determine the seawater samples collected 446–1316 km off the FDNPP accident site in the Northwest Pacific in July of 2013. The obtained 239+240Pu activity concentrations of 1.21–2.19 mBq/m3 and the 240Pu/239Pu atom ratios of 0.198–0.322 suggested that there was no significant Pu contamination from the accident to the Northwest Pacific.
Similar content being viewed by others
Introduction
Plutonium (Pu) is present in the marine environment mainly as a result of human activities related to atmospheric nuclear weapon tests, nuclear fuel reprocessing and nuclear accidents. From the viewpoint of radio-toxicity and long-term radiation effects to humans, Pu is by far one of the most important transuranic elements that have been released into the environment from nuclear plant accidents1. The element has twenty isotopes with mass ranging from 228 to 247. Among them, 240Pu and 239Pu are the most important due to their long half-lives(6537 y and 24100 y, respectively) and high abundance. As the result, 239+240Pu activity concentration can serve as an excellent tracer for studying sedimentary processes2,3, scavenging processes4, ocean current pathway5, particle transportation6 and other oceanic processes7. Moreover, 240Pu/239Pu atom ratio is of great interest because it has been well characterized for various sources, applied widely as a fingerprint to identify radioactive contamination sources in marine and terrestrial environments6,7. For instance, 240Pu/239Pu atom ratio of weapons-grade Pu ranges from 0.02 to 0.05, while nuclear reactor-grade 240Pu/239Pu ranges from 0.2 to 1.0 depending on irradiation conditions of the fuel8. The worldwide integrated global fallout 240Pu/239Pu is characterized by ratios of 0.17–0.19 with the average of 0.176 ± 0.0149. While the Pacific Proving Grounds (PPG) derived 240Pu/239Pu atom ratio is greater than 0.3010, which is close to that from the Fukushima Dai-ichi Nuclear Power Plant (FDNNP) with the value of 0.303–0.33311,12.
Approximately 15 PBq of 239+240Pu were released into the environment from atmospheric weapon tests conducted during the period of the 1950 s and 1960 s, and a few GBq of Pu were released to the marine environment from fuel reprocessing plants up until the present time1. It is reported that the total amount of 239+240Pu released into the ocean was ~12 PBq by the year 200013,14. For the Pacific Ocean, it was estimated that total 239+240Pu deposited from the atmospheric nuclear weapon testing is around 8.6 PBq, which included 5.1 PBq of 239+240Pu in local and regional fallout14. The total seawater volume is ~1.37 × 1018 m3 15, therefore, the resulting concentration of 239+240Pu in seawater is extremely low (~mBq/m3 or ~fg/L level). Moreover, the decreasing trend of 239+240Pu activity concentration with an apparent half-residence time of 5–21 y in surface seawater of the world’s oceans since 1970 is still unchanged16,17,18,19. The 239+240Pu activity concentrations in the surface seawater of the North Pacific decreased more than ten-fold from the early 1970 s (8.1–35 mBq/m3) to the year 2000 (0.3–2.7 mBq/m3)16. This makes measuring the current activity concentration of Pu in seawater very challenging. If the seawater samples were measured by a conventional radiometric analysis technique such as alpha-spectrometry, which counts the “dying” Pu atoms, a large volume of seawater (~200 L) and long analysis times (1–2 weeks) are typically needed. Meanwhile, alpha-spectrometry has the disadvantage of lacking the capability to provide the 240Pu/239Pu atom ratio, since these isotopes have similar alpha particle energies. By contrast, mass spectrometry, an atom-counting technique, has several advantages. It can be utilized with a small sample volume (several to tens of liters), simple sample preparation, short detection time, high sensitivity and precision as well as availability of accurate isotope ratio information, which is ideal for the determination of Pu isotopes in seawater.
Various analytical methods based on mass spectrometry have been developed including thermal ionization mass spectrometer (TIMS), accelerator mass spectrometer (AMS) and inductively coupled plasma mass spectrometry (ICP-MS)1,20,21. Among the mass spectrometric methods, ICP-MS is one of the most frequently employed for the last decade22. In recent years, the evolution of a new SF-ICP-MS with Jet Interface has developed for ultra-trace determination of Pu from femtogram (10−15 g) level to attogram (10−18 g) level. This allows comparable or even better sensitivity and detection limit than AMS23, which is ideal for the rapid determination of Pu isotopes in small volume seawater samples.
Even when the highly sensitive SF-ICP-MS is applied to determine Pu isotopes in seawater samples, the plutonium in the small volume seawater samples needs to be pre-concentrated. In addition, due to the serious mass interferences caused by the peak tailing effect of 238U+ (3.3 ng/ml in seawater, 9–10 orders of magnitude higher than that of Pu) and formation of uranium hydrides (238UH+ and 238UH2+), the 238U concentration in the final sample solution generally needs to be reduced to less than 5 pg/ml. To accomplish this removal of 238U, the U decontamination factor greater than ~107 is needed8,24. Therefore, the analytical procedure such as co-precipitation, anion-exchange chromatography, extraction chromatography and the combination of these methods have been taken in recent years for the determination of Pu in seawater24,25.
The FDNPP accident released a great amount of artificial radionuclides into the Pacific Ocean26. It is reported that 1.0–2.4 GBq of 239+240Pu was released into the terrestrial environment11,12. As considerable amounts of highly contaminated effluents originating from the inner structure of the reactor have been emitted into the ocean26,27, addition Pu isotopes derived by FDNPP accident might have entered into the Pacific Ocean. It is necessary and important to continue monitoring the Pu isotopes in seawater for long-term radiological assessment. However, many current analytical techniques for Pu isotopes in seawater are inadequate, not only due to the difficulties in sampling large volumes of seawater, but also due to the complexities of the chemical treatments required. In addition, many current analytical methods cause quite lot of waste acids enhancing great burdens for laboratory management. Therefore, a rapid analytical method of small-volume seawater samples is highly desired not only for emergency response and assessment, but also for improving sample throughput and reducing environmental hazards in routine analyses. In this work, a new analytical method to rapidly determine Pu isotopes in seawater by SF-ICP-MS including a Fe(OH)2 primary co-precipitation, CaF2/LaF3 secondary co-precipitation and TEVA+UTEVA+DGA extraction chromatographic separation was established. And the activity concentrations of 239+240Pu isotopes as well as 240Pu/239Pu atom ratios in the surface water of Northwest Pacific were determined based on this new method to assess the impacts of the Fukushima derived Pu isotopes on the marine environment.
Materials and Methods
Reagents and seawater samples
A Millipore Milli-Q-Plus water purification system was used for the preparation of high-purity water (>18.2 MΩ/cm). HNO3 (60–61%), HCl (35.0–37.0%), HF (49.5–50.5%), NH3H2O (25.0–27.9%), TiCl3 (20%), FeCl3·6H2O, C6H8O6 (VC, Ascorbic acid), NaNO2, and H3BO3 are of analytical grade and obtained from Kanto Chemical Co., INC, Japan. Ca(NO3)2·4H2O and La(NO3)2·6H2O are also of analytical grade purchased from by Wako Pure Chemical Industries, Ltd, Japan. Analytical-grade Iron (II) sulfamate (38–42%) aqueous solution are obtained from Strem Chemicals, USA. Ultrapure-grade HNO3 (68%) is used for sample preparation prior to ICP-MS measurement, which is purchased from Tama Chemicals, Japan. Three kinds of extraction resins TEVA, UTEVA and DGA (Eichrom Technologies, LLC) used in this study were 2 mL cartridges with grain sizes of 50–100 μm. 242Pu (CRM 130, plutonium spike assay and isotopic standard, New Brunswick Laboratory, USA), as a yield tracer, was used to spike the seawater samples. IAEA-443 certified seawater reference material was obtained from the International Atomic Energy Agency (IAEA, Vienna, Austria).
Surface seawater samples (~17 L for each sample collected from 0–1 m from the surface) were collected from 446–1316 km off the FDNNP site in the Northwest Pacific during the cruises of MR 13–04 in July 2013, which were used to assess the impact of Fukushima derived Pu isotopes on seawater. After sampling, seawater samples were acidified to pH ~2 with concentrated HCl and then filtered into a HDPE bucket (20 L) (Teraoka Company, Japan). Then the seawater samples were transferred to the land-based laboratory.
Seawater sample preparation
Prior to the analysis by SF-ICP-MS, seawater samples need a series of separation and purification to remove the matrix and interfering elements. The overall procedure is shown in Fig. 1.

Flow chart of the analytical procedure for the determination of Pu isotopes in seawater by extraction chromatography and SF-ICP-MS
Fe(OH) 2 co-precipitation
Transfer ca. 15 L of pre-filtered seawater into a plastic container. TiCl3 (0.5 mL of 20% per liter seawater) and 125 mg Fe3+ (5 mL of 25 mg/mL) were added to the sample under stirring in sequence. Fe was used as a carrier, and TiCl3 was used to reduce Pu (V, VI) to Pu (III) for complete co-precipitation of Pu with iron hydroxide. The concentrated NH3H2O was added to adjust pH = 8.8–9.0 for Fe(OH)2 co-precipitation under stirring. The formed Fe(OH)2 co-precipitates were stirred at least 30 mins by stirrers and then allowed to settle for about 4 hours. The sample supernatant were carefully siphoned and pumped away avoiding loss of Fe(OH)2 co-precipitates. The obtained Fe precipitate slurry (ca. 400 mL) was collected in two 250 mL centrifuge tubes and the small volumes (ca. 50 mL) of pure water were used to rinse the sample bucket, which was also combined into the 250 mL centrifuge tubes. The slurry was centrifuged under 3000 rpm for 15 min. After pouring out the supernatant, 20 mL of concentrated HNO3 and 0.57 pg of 242Pu spike were added into the first centrifuge tube to dissolve the precipitate, and then the obtained solution was transferred into the second centrifuge tube to dissolve the rest of precipitate. Rinse the first centrifuge tube with 5 mL of deionized water and pour the rinsing water into the second centrifuge tube. The final solution was ca. 3.4 M HNO3 solution with the volume of ca. 75 mL.
CaF 2 /LaF 3 co-precipitation
In order to form CaF2/LaF3 co-precipitation, 100 mg Ca2+, 100 mg La3+ and 7 mL of concentrated HF were added into the second centrifuge tube, followed by vigorous shaking and 15-minute settling. After centrifugation under 3000 rpm for 15 min, the supernatant was discarded and the precipitate was dissolved by 20 mL of 3 M HNO3 with the addition of 0.5 g of H3BO3. The solution was transferred into the 50 mL centrifuge tube and Pu (III) was adjusted to Pu (IV) by the addition of 0.3 g of NaNO2 and heated at 40 °C for 0.5 h in a temperature–controllable heating apparatus (DigiPREP Jr, SCP SCIENCE, Canada).
Separation and purification
As shown in Fig. 1, the sample solution was loaded onto a TEVA resin cartridge (at a flow rate of 1 drop per second) which had been preconditioned with 10 mL of 3 M HNO3. An additional 10 mL of 3 M HNO3 was used to rinse the 50 ml centrifuge tube and leached through the TEVA resin cartridge to remove Ca, Fe, and rare earth elements (REEs), followed by 40 mL of 1 M HNO3 to remove U, Pb, and Tl, and 10 mL of 9 M HCl to remove Th, and Bi (at a flow rate of 2 drops per second). Before the elution of Pu, an UTEVA+ DGA resin cartridge (also preconditioned by 10 mL of 3 M HNO3) was connected to the bottom of the TEVA resin cartridge. Then 20 mL of 3 M HNO3 −0.1 M ascorbic acid −0.02 M Fe2+ (iron(II) sulfamate) was employed to reduce Pu (IV) to Pu (III) and elute Pu (III) from TEVA resin (1 drop per second) to the DGA resin. Then, the TEVA and UTEVA resin cartridges were removed. The DGA resin cartridge was rinsed by 30 mL of 0.1 M HNO3 to remove U, Tl, Pb, and Fe (2 drops per second). Next, the plutonium on the DGA resin was eluted into a 50 mL PTFE beaker by 20 mL of 0.5 M HCl −0.1 M NH2OH·HCl (1 drop per second). The eluted sample solution was evaporated to dryness on a hot plate with the temperature of 250 °C and dissolved using 4 mL of aqua regia. After heating the dissolved sample solution to dryness on a hot plate with the temperature of 200 °C, 1 mL of ultrapure HNO3 was added and heated to near dryness at 200 °C. Finally, the sample was dissolved in 0.7 mL of 4% HNO3 and ready for SF-ICP-MS measurement.
SF-ICP-MS measurements
The measurement of Pu isotopes was carried out using a SF-ICP-MS (Element XR, Thermo Fisher Scientific Inc., Germany). An APEX-Q high efficiency sample introduction system (Elemental Scientific Inc, USA) combined with a membrane desolvation unit (ACM) and equipped with a conical concentric nebulizer (~0.15 mL/min) was used for sample introduction. A 238U standard solution (0.02 ng/mL) was used to adjust optimum performance daily before sample detection. The low resolution mode (m/Δm = 400) was used in order to utilize the maximum instrument sensitivity. The instrument and data acquisition settings of APEX-Q/SF-ICP-MS were detailed elsewhere23.
Results and Discussion
Pu valence adjustment and co-precipitation
The dissolved Pu isotopes in seawater are present primarily as Pu (V) or Pu (VI), which do not undergo co-precipitation as favorably as Pu (III) and Pu (IV). Therefore converting Pu to a reduced oxidation state is necessary to enhance the efficiency of co-precipitation1. Among the specific reducing agents NH3OHCl, Na2SO3, K2S2O5 and TiCl328,29,30, TiCl3 was demonstrated having not only the ability of enhancing chemical recovery but also the advantage of being removed in the subsequent fluoride co-precipitation step25. As for the primary co-precipitation of Pu isotopes in seawater, MnO2 and Fe(OH)3 methods are frequently employed25,28,29. Compared to Fe(OH)3, which can be easily removed in the subsequent fluoride removal step, the removal of Mn from the final precipitates is somewhat more troublesome25,31. In addition, smaller amounts of reagents are required when applying the Fe(OH)3 co-precipitation compared with the MnO2 method. Actually, during the Fe(OH)3-primary co-precipitation in our method, TiCl3 reduces Fe3+ to Fe2+. Hence, the final co-precipitate here is Fe(OH)2.
For the need of further concentration, secondary co-precipitation is necessary. For the small volume of samples, rare-earth fluoride methods are extremely suitable for the co-precipitation of Pu and Pu separated from a supernatant solution containing U, Pt, Fe, Cr, etc.32,33. It was reported that LaF3 or NdF3 can selectively co-precipitate Pu (III, IV) effectively21,25,34. Another advantage of CaF2/LaF3 co-precipitation is that 60% of U can be removed by this procedure35. Besides, the fluoride co-precipitation can also remove the large amount of iron and titanium used in the primary co-precipitation step25. Therefore, in this study, CaF2/LaF3 was selected to for the secondary co-precipitation.
Pu concentration efficiency of Fe(OH) 2 co-precipitation
242Pu is one of nuclear fuel nuclides. There are very strict management rules for the use of nuclear fuel nuclides in Japan36. It is controlled in some specified area and not allowed to take to the laboratory onboard, which limits its use for tracing Pu in-site pre-concentration on board and enhance the burden of sample transportation. In order to solve this problem, we attempted to study the feasibility of adding 242Pu spike after the primary Fe(OH)2 co-precipitation rather than at the beginning of experiment into the seawater samples directly. To this end, a set of experiments were designed to estimate the Pu concentration efficiency (CE) (ratio of Pu amount in the Fe(OH)2 co-precipitation to that in the original seawater sample) of Fe(OH)2 co-precipitation and its stability. First, we used primary co-precipitation procedure in Fig. 1 to remove the original 239+240Pu (unknown amount) from seawater and keeping the supernatant to get Pu-free seawater sample (15–16.6 L). Second, we added the 2.5–20 mL of IAEA-443 standard seawater into the Pu-free seawater sample to obtain seawater samples with very accurate known 239+240Pu amounts, which can make sure that we get the accurate recovery more scientifically. After the primary co-precipitation with Fe(OH)2, 0.57 pg of 242Pu spike were added into the solution before the CaF2/LaF3 co-precipitation. So the final results of 239+240Pu we calculated represented the 239+240Pu activity concentration of the IAEA-443 standard added seawater without an estimation of Pu recovery during Fe(OH)2 co-precipitation. If the value was divided by the certified 239+240Pu activity concentration of added IAEA-443 reference seawater, the Pu CE of Fe(OH)2 co-precipitation can be estimated. As shown in Table 1, Pu CE of Fe(OH)2 co-precipitation ranged from 98.8% to 103% with an average of 101 ± 2% suggesting that Fe(OH)2 co-precipitation can quantitatively recover Pu isotopes in seawater. In addition, all of the CEs were consistent in the range of 2σ standard deviation (Table 1), which demonstrated its reliable stability for concentrating Pu isotopes from seawater. In other words, the loss of Pu during Fe(OH)2 co-precipitation is negligible. So our results confirmed that this method is practical.
As a matter of fact, in addition to the advantage of in–site processing seawater samples, there is another advantage of our method. It can greatly reduce the radioactive wastes that contains 242Pu and the resulting burden of waste disposal for laboratories with restrictions on the use of nuclear fuel nuclides.
Pu chemical recovery and decontamination factor of U
Chemical separations of Pu are commonly carried out using anion exchange resins or extraction chromatography. Anion-exchange resins such as AG 1 × 8, AG MP-1M and Dowex 1 × 8 have attracted much attention for their low cost and wide applicability as well as strong tolerance capacity for matrix elements1,22. On the other hand, extraction resins such as TEVA, UTEVA, TRU and DGA have the advantages of short sample processing time, high recovery and less amount of acidic wastes. It is worth noting that, the prerequisite for using extraction resins for Pu separation is that large amounts of matrix elements must be removed almost completely through suitable pretreatment22. TEVA resin has a high capacity factor for Pu (3 × 104) and is more effective for samples containing high concentrations of Fe, Mn, Ce, and interfering elements, such as U, Pb, Hg, which become the first choice for Pu separation8,35. UTEVA resin has much higher selectivity for U8. DGA resin can retain both trivalent and tetravalent actinides and further remove U, Tl, Pb, and Fe34,35. It is reported that the decontamination factor (DF) of U up to ~3 × 106 can be achieved for the urine sample when TEVA+UTEVA+DGA resin combination was used25. It met the requirements of U DF for Pu determination in seawater. Therefore, based on the reason abovementioned, we choose the TEVA+UTEVA+DGA resins combination for Pu separation in seawater in this study.
Pu chemical recovery is one of the crucial indicators to assess the effectiveness of a Pu determination method. A high recovery is essential for the determination of Pu in small volume seawater sample, because the amount of Pu in seawater samples is not much higher than the detection limit of the SF-ICP-MS. It was found that the signal intensity of 240Pu atom was just around 50 ~ 80 cps when we measured the surface seawater (15–20 L) of Northwest Pacific, which suggested that the small fluctuation of recovery might result in a large influence on the accuracy and precision of the result. Lower recovery could result in a failure to analyze the 239+240Pu activity concentration and 240Pu/239Pu atom ratio. In addition to Pu chemical recovery, the DF of U is another crucial indicator to assess the Pu determination method. For the ultra-trace analysis of Pu isotopes, even though there is only a micro interference of uranium, it will hinder accuracy and precision of the Pu activity concentrations and their atom ratios.
To better understand the Pu chemical recovery and DF of U of our method, we repeated the Pu measurement (n = 8) for the same seawater sample collected from the Northwest Pacific. The seawater sample (120 L) were divided into 8 subsamples with the volume of 15 L each. The results are shown in Table 2. Pu chemical recovery of this method is 74–88% with a mean of 83 ± 5% (n = 8). U DF is 1.6 × 106 ~ 2.4 × 107 with a mean of (1.5 ± 0.9) × 107. Table 3 lists the comparison of our method with the reported analytical methods of seawater including Pu chemical recovery, DF of U, limit of detection (LOD) and so on. As shown in Table 3, Pu chemical recovery of this method was comparable to that of the other Pu analysis methods of seawater, which suggested that the satisfactory chemical recovery was achieved in our new method.
As shown in Table 2, U DFs of our method (1.6 × 106 ~ 2.4 × 107 with the mean of 1.5 × 107 ± 0.9 × 107) are better than that of the most previously reported values in Table 3. As mentioned in the introduction section, a U DF of ~107 means that Pu was successfully separated from U. Understandably, the 238U signal intensities in the final Pu fraction for the seawater sample detected by SF-ICP-MS were about 100,000 cps, similar to that of the operational blank. As the 238UH+/238U+ ratio for our SF-ICP-MS system was less than 3 × 10−5, the contribution of 238UH+ to the 239Pu+ of interest was below 3 cps. Considering that the intensity of 239Pu in the Northwest Pacific seawater samples (15–20 L, ca. 1 mBq/m3 for 239+240Pu) exceeded 230 cps, our method made the U interference negligible for determination of Pu isotopes in seawater.
Accuracy, precision and reproducibility of the analytical method
Accuracy and precision are the two essential indicators for the reliability of one analytical method. Figure 2 plots the data of samples in Table 1. It presented the measured results of IAEA 443-spiked seawater samples with different activity concentrations of 239+240Pu and the relationship between the precisions of single measurement of 240Pu/239Pu atom ratio with a serial 239+240Pu activity concentrations (2.65–18.55 mBq/m3). The mean of measured 240Pu/239Pu atom ratios (n = 8) was 0.235 ± 0.010 (k = 2). Their precision (RSD %) and accuracy were 2.2% and 3.0%, respectively. The single measurement precision increased with increasing 239+240Pu activity concentrations ranging from 4.0% to 16.9%. However, the 240Pu/239Pu atom ratios remain within the 95% confidence level of the overall average, suggesting that this method is a suitable technique for accurate measurements of Pu isotopic ratio in seawater sample with ultra-trace concentrations of Pu. As for the precision of single measurement of seawater 239+240Pu activity concentrations (2.65–18.55 mBq/m3), they ranged from 4.9–11.4%.

Accuracy and precision of 240Pu/239Pu atom ratio measurement obtained from serial IAEA-443 spiked samples. The error bars represent measuring error of each analysis. Horizontal solid and dashed lines represent the overall average 240Pu/239Pu atom ratios and expanded standard uncertainties (k = 2), respectively.
We also evaluated the reproducibility of the entire analytical method by repeated measurements (n = 8) of 239+240Pu activity concentration and 240Pu/239Pu atom ratio in a seawater sample with a typical 239+240Pu activity concentration <2 mBq/m3 in Northwest Pacific. As shown in Fig. 3, for the 8 repeated measurements, the mean of 239+240Pu activity concentration was 1.86 ± 0.17 mBq/m3 (k = 2) and the mean of 240Pu/239Pu atom ratios was 0.245 ± 0.023 (k = 2). The precision of 8-time repeated measurement of 240Pu/239Pu atom ratios and 239+240Pu activity concentrations were 4.7% and 4.6%, respectively. The single measurement precision of the 240Pu/239Pu atom ratios and 239+240Pu activity concentrations ranged from 11.7–17.5% and 11.5–17.7%, respectively. All the single measurement results of 239+240Pu activity concentration and 240Pu/239Pu atom ratio varied within 2σ of the mean values. It suggested that this method can achieve a precision of <5% for both 240Pu/239Pu atom ratios and 239+240Pu activity concentrations when 15 L of seawater samples with the typical 239+240Pu activity concentration of the surface seawater in Northwest Pacific (~1.9 mBq/m3, corresponding to 9.8 fg/mL 239Pu and 2.2 fg/mL 240Pu in the final measured solution) are measured. In addition, as presented in Table 2, the Pu recovery ranged from 74% to 88%, with a mean of 83% ± 5%, indicating the analytical method is highly stable and reproducible. A detailed comparison with other published analytical methods for seawater Pu analysis is presented in Table 3. As shown in Table 3, the precision and accuracy for both 239+240Pu activity and 240Pu/239Pu atom ratio of our new method is comparable or better than most of the published methods.

Precision of 240Pu/239Pu atom ratios (a) and 239+240Pu activity concentrations (b) measurement obtained from the Northwest Pacific seawater samples. The error bars represent measuring error of each analysis. Horizontal solid and dashed lines represent the overall average values and expanded standard uncertainties (k = 2), respectively.
Limit of detection and validation of the method
High sensitivity and low detection limit are needed for the high performance analysis of Pu in small-volume seawater due to the low concentration of Pu. We used the APEX-Q sample introduction system to promote the sensitivity of SF-ICP-MS (Element XR). As described in previous work23, the sensitivity of the whole system can be improved up to 60Mcps/ppb, which can reduce greatly the volume of the samples.
The instrument detection limits of 239Pu and 240Pu were determined based on the estimation of 3 times the standard deviation of a 4% HNO3 blank solution. Similarly, the LODs of our method were calculated according to 3 times the standard deviation of the operation blanks. On the basis of analyzing 15 L of pure water with a Pu recovery of 83%, the LODs for 239Pu and 240Pu were both 0.08 fg/mL, corresponding to 0.01 mBq/m3 for 239Pu and 0.05 mBq/m3 for 240Pu when a 15 L of seawater was measured. As shown in Table 3, the LODs of this study is much lower than most of the reported LODs and comparable with Bu et al.24 but higher than that in the work reported by Lindahl et al.8. It should be noted that Lindahl et al.8 used 1% HNO3 solution to estimate the LODs. Their LODs were merely the MC-ICP-MS instrumental LODs, not the real LODs of entire analytical method. Therefore, the LODs of this study are among the lowest in the reported LODs.
The seawater reference material IAEA-443 was employed to validate this new method. A serial of IAEA-443 spiked seawater samples (2.5–20 mL of IAEA-443 in 15–16.6 L “Pu pre-removed” seawater) were used to illustrate the validation of our method. The results of 240Pu/239Pu atom ratios ranged from 0.225 ± 0.009 to 0.241 ± 0.040, which agreed well with the certificate values of 0.229 ± 0.006 within the range of error (Table 1). As for the 239+240Pu activity concentrations, each of them agreed well with the corresponding certificate value within the range of error (Table 1).
Sample throughput
The complete analytical method takes about 12 h: Fe(OH)2 co-precipitation, 5 h; CaF2/LaF3 co-precipitation, 1 h; Pu separation on extraction resin, 2 h; and sample preparation for SF-ICP-MS measurements, 4 h. Compared to analytical methods employed conventional ion-exchange chromatographic separation, which usually takes about 3–5 days for Pu separation, the new method in this study significantly shortens the analytical time. If the vacuum box with 24 positions were used, 24 samples could be separated and purified simultaneously. It would be an extremely high sample throughput. In addition, this method produces less amount of hazardous acidic wastes and requires less evaporation of acids, greatly reduces the burden of radioactive laboratory management.
Assessment on the Impact of Fukushima-derived Pu isotopes on seawater
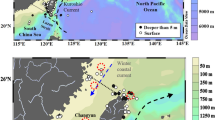
The developed method was applied to determine the 239+240Pu activity concentrations and 240Pu/239Pu atom ratios in 16 surface seawater samples obtained 446–1316 km off the FDNPP site in the Northwest Pacific in July 2013 (Fig. 4). The results are shown in Table 4 with other relevant oceanography information. The 239+240Pu activity concentrations ranged from 1.21 ± 0.18 to 2.19 ± 0.23 mBq/m3, showing that the value was within the range of pre-FDNPP accident period16. For 240Pu/239Pu atom ratios, values were from 0.198 ± 0.026 to 0.322 ± 0.042, respectively, also showing that they were within the range of the Northwest Pacific in pre-FDNPP accident period37. There were no obviously relationships of 239+240Pu activity concentrations and 240Pu/239Pu atom ratios with salinities (Fig. 5).

Surface seawater sampling stations in the Northwest Pacific in July, 2013. This figure was drawn using Surfer version 12.5.905 (http://www.goldensoftware.com).

Relationships of 239+240Pu activity concentrations and 240Pu/239Pu atom ratios with salinities.
After the FDNPP accident, 239+240Pu activity concentrations and 240Pu/239Pu atom ratios in seawater of Northwest Pacific were investigated to study if there was significant impact for Pu isotopes in seawater before and after the FDNPP accident. The seawater zone of <30 km, 30–200 km and >200 km off FDNNP were investigated respectively19,24,38,39,40. Their results suggested no significant Pu contamination from the accident in these areas. The 240Pu/239Pu atom ratios (0.198–0.322) were higher than the global fallout ratio of 0.176 ± 0.0149, but lower than that of the PPG nuclear weapon tests of 0.30–0.3641,42,43. Coincidentally, the FDNNP derived 240Pu/239Pu atom ratios had the similar range of 0.30–0.33 with that of PPG nuclear weapon tests11. Thus we could not conclude whether the FDNPP accident derived Pu isotopes had contributions to the 16 measured samples based on 240Pu/239Pu atom ratio in this study. Furthermore, 241Pu/239Pu atom ratios in the seawater should be measured because the FDNNP derived Pu isotopes had another characteristic 241Pu/239Pu atom ratios of 0.103–0.13511, which had much higher 241Pu/239Pu atom ratios compared to that of global fallout (0.00089) and PPG nuclear weapon tests (0.0017–0.0024)38,39,44,45. However, it is still difficult to measure the 241Pu in seawater at present. The relative technique remains to be further developed. However, the comparison of 239+240Pu concentrations before and after the FDNPP accident can be made to confirm whether it is above the range of that before FDNPP accident. Oikawa et al.18 reported the data of Pu isotopes in the surface seawater of the sites of commercial nuclear power stations around the Japanese Island from 2008 to 2010. In addition, the IAEA-MARiS-Maine Information System also records the variation of Pu isotopes in surface seawater of the Western North Pacific (15–40°N, 110–160°E) from 1966 to 200346. All these data could be used as the baseline data before the FDNPP accident. The 239+240Pu activity concentrations of the zone of >400 km from the FDNPP in this study (1.21–2.19 mBq/m3) was typically in the range of the background data of the Northwest Pacific (Fig. 6)18,19,24,46, indicating negligible Pu contamination from the accident.

Comparison of 239+240Pu activities in this study with the historical 239+240Pu data in surface seawater of the Western North Pacific. (The data of area 15–40°N, 110–160°E in 1966–2003 are from IAEA-MARIS-Marine Information System46; the data of the commercial nuclear power station sites around the Japanese Island from 2008 to 2010 are from Oikawa et al.18.
Wu et al.47 confirmed that even 60 years after the 1950 s, the PPG was still a dominant Pu source in the marginal seas of the Northwestern Pacific due to continuous transport of remobilized Pu from the Marshall Islands along the North Equatorial Current followed by the transport of the Kuroshio Current and its extension (Fig. 4). Now that abovementioned work has proved that there were negligible Pu isotopes released from the FDNPP accident. Therefore, that the 240Pu/239Pu atom ratios of this study were higher than the global fallout but lower than that of the PPG nuclear weapon tests suggested that the Pu isotopes mainly come from global fallout and PPG. We can estimate the contributions of global fallout and PPG close-in fallout Pu using the two end-member mixing model proposed by Krey et al.9:
where R refers to 240Pu/239Pu ratio, subscripts P, G and S refer the PPG close-in fallout, and the global stratospheric fallout and the samples measured in this study, respectively. The value 3.674 is the ratio of the specific activity of 240Pu to 239Pu, by which the atom ratio is converted to the activity ratio. Krey et al.9 reported the 240Pu/239Pu atom ratio of global fallout to be 0.18 ± 0.02 between 30°N and 60°N. The close-in fallout in the PPG had a 240Pu/239Pu atom ratio of 0.363 ± 0.00448. The contributions of the PPG close-in fallout of this study were with the range of 13%-84% (Table 4). Buesseler41 suggested that Pu derived from the PPG would be preferentially removed from the water column, compared with the global stratospheric fallout Pu that is more soluble. Therefore the high contributions of PPG close-in fallout Pu (e.g. PPG contributions of 84% with the 240Pu/239Pu atom ratio of 0.322) in this study might suggest that the latest transport of the PPG-derived Pu to the corresponding area.
Summary
In this study, a high-performance method for the determination of Pu isotopes in small volume seawater based on Fe(OH)2 pre-concentration, CaF2/LaF3 co-precipitation, TEVA+UTEVA+DGA extraction chromatographic separation and SF-ICP-MS measurement was reported. The Pu concentration efficiency of Fe(OH)2 co-precipitation was close to ~100%. Pu chemical recovery of the developed method ranged from 74–88% with the mean of 83 ± 5%. A high U decontamination factor of ~107 was achieved, which made the U interference negligible for the determination of ultra-trace Pu isotopes. The LOD for 239Pu and 240Pu were both 0.08 fg/mL, corresponding to 0.01 mBq/m3 for 239Pu and 0.05 mBq/m3 for 240Pu when a 15 L volume of seawater was measured. The entire analytical method only took about 12 h. This method was applied to determine the seawater samples collected 446–1316 km off the FDNPP accident site in the Northwest Pacific in July of 2013. The 239+240Pu activity concentrations of 1.21–2.19 mBq/m3 and the 240Pu/239Pu atom ratios of 0.198–0.322 were obtained, which also suggested there was no significant Pu contamination from the accident to the Northwest Pacific.
References
Kim, C. S., Kim, C. K., Martin, P. & Sansone, U. Determination of Pu isotope concentrations and isotope ratio by inductively coupled plasma mass spectrometry: a review of analytical methodology. J. Anal. At. Spectrom. 22, 827–841 (2007).
Liu, Z. et al. Pu and 137Cs in the Yangtze River Estuary sediments: distribution and source identification. Environ. Sci. Technol. 45(5), 1805–1811 (2011).
Pan, S. M., Tims, S. G., Liu, X. Y. & Fifield, L. K. 137Cs, 239+240Pu concentrations and the 240Pu/239Pu atom ratio in a sediment core from the sub-aqueous delta of Yangtze River estuary. J. Environ. Radioact. 102(10), 930–936 (2011).
Hirose, K. Complexation-scavenging of plutonium in the ocean. Radioprotection 32, C2.225–C2.230 (1997).
Huh, C. A. et al. Natural radionuclides and plutonium in sediments from the western Arctic Ocean: Sedimentation rates and pathways of radionuclides. Deep Sea Res. II. 44(8), 1725–1743 (1997).
Zheng, J. & Yamada, M. Plutonium isotopes in settling particles: transport and scavenging of Pu in the western Northwest Pacific. Environ. Sci. Technol. 40(13), 4103–4108 (2006).
Yamada, M. & Zheng, J. 239Pu and 240Pu inventories and 240Pu/239Pu atom ratios in the equatorial Pacific Ocean water column. Sci. Total Environ. 430, 20–27 (2012).
Lindahl, P. et al. Ultra-trace determination of plutonium in marine samples using multi-collector inductively coupled plasma mass spectrometry. Anal. Chim. Acta. 671, 61–69 (2010).
Krey, P. W. et al. Mass isotopic composition of global fall-out plutonium in soil in Transuranium Nuclides in the Environment (ed. Horn, W.) 671–678 (IAEA, 1976).
Lindahl, P. et al. Spatial and temporal distribution of Pu in the Northwest Pacific Ocean using modern coral archives. Environ. Int. 40, 196–201 (2012).
Zheng, J. et al. Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident. Sci. Rep. 2, 304 (2012).
Zheng, J., Tagami, K. & Uchida, S. Release of plutonium isotopes into the environment from the Fukushima Daiichi Nuclear Power Plant Accident: what is known and what needs to be known. Environ. Sci. Technol. 47(17), 9584–9595 (2013).
Aarkrog, A. Input of anthropogenic radionuclides into the World Ocean. Deep-Sea Res. II 50(17–21), 2597–2606 (2003).
Hamilton, T. F. Linking legacies of the Cold War to arrival of anthropogenic radionuclides in the oceans through the 20th century in Radioactivity Concentration in the Environment (ed. Livingston, H.D) 23–78 (Elsevier, 2004).
Chronological Scientific Tables (ed. National Astronomical Observatory, Japan) (Maruzen, 2016).
Hirose, K. & Aoyama, M. Analysis of 137Cs and 239,240Pu concentrations in surface waters of the Pacific Ocean. Deep Sea Res. II 50(17), 2675–2700 (2003).
Oikawa, S. et al. Plutonium isotopes concentration in seawater and bottom sediment off the Pacific coast of Aomori sea area during 1991–2005. J. Environ. Radioact. 102(3), 302–310 (2011).
Oikawa, S., Watabe, T. & Takata, H. Distributions of Pu isotopes in seawater and bottom sediments in the coast of the Japanese archipelago before and soon after the Fukushima Dai-ichi Nuclear Power Station accident. J. Environ. Radioact. 142, 113–123 (2015).
Bu, W. et al. Pu distribution in seawater in the near coastal area off Fukushima after the Fukushima Daiichi Nuclear Power Plant accident. J. Nucl. Radiochem. Sci. 15(1), 1–6 (2015).
Lee, S. H. et al. Analysis of plutonium isotopes in marine samples by radiometric, ICP-MS and AMS techniques. J. Radioanal. Nucl. Chem. 248(3), 757–764 (2001).
Buesseler, K. O. & Justin, E. H. The mass spectrometric determination of fallout 239Pu and 240Pu in marine samples. J. Environ. Radioact. 5, 425–444 (1987).
Cao, L. G. et al. Plutonium determination in seawater by inductively coupled plasma mass spectrometry: A review. Talanta 151, 30–41 (2016).
Zheng, J. Evaluation of a new sector-field ICP-MS with Jet Interface for ultra-trace determination of Pu isotopes: from femtogram to attogram levels. J. Nucl. Radiochem.Sci. 15(1), 7–13 (2015).
Bu, W. et al. Ultra-trace plutonium determination in small volume seawater by sector field inductively coupled plasma mass spectrometry with application to Fukushima seawater samples. J. Chromatogr. A. 1337, 171–178 (2014).
Maxwell, S. L. et al. Rapid determination of actinides in seawater samples. J. Radioanal. Nucl. Chem. 300(3), 1175–1189 (2014).
Buesseler, K. et al. Fukushima Daiichi Derived Radionuclides in the Ocean: Transport, Fate, and Impacts. Annu. Rev. Mar. Sci. 9, 173–203 (2017).
IAEA. The Fukushima Daiichi Accident, technical volume 1/5, description and context of the accident http://www-pub.iaea.org/MTCD/Publications/PDF/AdditionalVolumes/P1710/Pub1710-TV1-Web.pdf (2015).
Qiao, J., Hou, X., Steier, P. & Golser, R. Sequential injection method for rapid and simultaneous determination of 236U, 237Np, and Pu isotopes in seawater. Anal. Chem. 85(22), 11026–11033 (2013).
Benkrid, M. & Noureddine, A. Plutonium isotopes concentration in seawater along the Algerian coast. Sci. Technol. Nucl. Ins. 2007, 1–5 (2007).
Sidhu, R. S. A robust procedure for the determination of plutonium and americium in seawater. J. Radioanal. Nucl. Chem. 256(3), 501–504 (2003).
Qiao, J., Hou, X., Miró, M. & Roos, P. Determination of plutonium isotopes in waters and environmental solids: A review. Anal. Chim. Acta. 652(1), 66–84 (2009).
Maxwell, S. L., Culligan, B. K., Warren, R. A. & McAlister, D. R. Rapid method for the determination of 226Ra in hydraulic fracturing wastewater samples. J. Radioanal. Nucl. Chem. 309, 1333–1340 (2016).
Maxwell, S. L. et al. Determination of 237Np and Pu isotopes in large soil samples by inductively coupled plasma mass spectrometry. Anal. Chim. Acta 682(1), 130–136 (2010).
Levy, P. P. et al. Marine anthropogenic radiotracers in the Southern Hemisphere: New sampling and analytical strategies. Prog. Ocean. 89(1), 120–133 (2011).
Wang, Z. T. et al. High-performance method for determination of Pu isotopes in soil and sediment samples by sector field-inductively coupled plasma mass spectrometry. Anal. Chem. 89, 2221–2226 (2017).
Act on the Regulation of Nuclear Source Material, Nuclear Fuel Material and Reactors. http://www.japaneselawtranslation.go.jp/law/detail/?id=1941&vm=03&re=01 (2009)
Zheng, J. et al. Distribution of Pu isotopes in marine sediments in the Pacific 30 km off Fukushima after the Fukushima Daiichi nuclear power plant accident. Geochem. J. 46, 361–369 (2012).
Sakaguchi, A. et al. Isotopic determination of U, Pu and Cs in environmental waters following the Fukushima Daiichi Nuclear Power Plant accident. Geochem. J. 46, 355–360 (2012).
Hain, K. et al. Plutonium Isotopes (239–241Pu) Dissolved in Pacific Ocean Waters Detected by Accelerator Mass Spectrometry: No Effects of the Fukushima Accident Observed. Environ. Sci. Technol. 51, 2031–2037 (2017).
Wendel, C. C. S. et al. No Fukushima Dai-ichi derived plutonium signal in marine sediments collected 1.5–57 km from the reactors. Appl. Radiat. Isot. 129, 180–184 (2017).
Buesseler, K. O. The isotopic signature of fallout plutonium in the North Pacific. J. Environ. Radioact. 36(1), 69–83 (1997).
Yamada, M., Zheng, J. & Wang, Z. L. 240Pu/239Pu atom ratios in seawater from Sagami Bay, western Northwest Pacific Ocean: sources and scavenging. Environ. Radioact. 98, 274–384 (2007).
Muramatsu, Y. et al. Measurement of 240Pu/239Pu isotopic ratios in soils from the Marshall Islands using ICP-MS. Sci. Total Environ. 278, 151–159 (2001).
Kelly, M., Bond, L. A. & Beasley, T. M. Global distribution of Pu isotopes and 237Np. Sci. Total Environ. 237–238, 483–500 (1999).
Lee, S.-H. et al. Distribution and inventories of 90Sr, 137Cs, 241Am and Pu isotopes in sediments of the Northwest Pacific Ocean. Mar. Geol. 216, 249–263 (2005).
Marine Information System of International Atomic Energy Agency (MARiS). https://maris.iaea.org/Home.aspx (2011).
Wu, J. et al. Isotopic composition and distribution of plutonium in Northern South China Sea sediments revealed continuous release and transport of Pu from the Marshall Islands. Environ. Sci. Technol. 48(6), 3136–3144 (2014).
Diamond, H. et al. Heavy isotope abundances in Mike thermonuclear device. Phys. Rev. 119, 2000–2004 (1960).
Norisuye, K. et al. Large volume preconcentration and purification for determining the 240Pu/239Pu isotopic ratio and 238Pu/239+240Pu alpha-activity concentration ratio in seawater. J. Radioanal. Nucl. Chem. 267(1), 183–193 (2006).
Chen, Q., Dahlgaard, H., Nielsen, S. P. & Aarkrog, A. 242Pu as tracer for simultaneous determination of 237Np and 239,240Pu in environmental samples. J. Radioanal. Nucl. Chem. 253(3), 451–458 (2002).
Kim, C. S., Kim, C. K. & Lee, K. J. Determination of Pu isotopes in seawater by an online sequential injection technique with sector field inductively coupled plasma mass spectrometry. Anal. Chem. 74(15), 3824–3832 (2002).
Qiao, J. et al. Rapid multisample analysis for simultaneous determination of anthropogenic radionuclides in marine environment. Environ. Sci. Technol. 48(7), 3935–3942 (2014).
Levy, I. et al. Marine anthropogenic radiotracers in the Southern Hemisphere: New sampling, and analytical strategies. Prog. Oceanogr. 89, 120–133 (2011).
Rosa, L. J. et al. Recent developments in the analysis of transuranics (Np, Pu, Am) in seawater. J. Radioanal. Nucl. Chem. 263(2), 427–436 (2005).
Hirose, K. & Sugimura, Y. A new method of plutonium speciation in sea water. J. Radioanal. Nucl. Chem. 92(2), 363–369 (1985).
Acknowledgements
We would like to thank the scientific parties, the Captains and crew of R/V MIRAI MR 13–04 cruises in July 2013 for their help in seawater sampling. This work was supported by the Grant of Fukushima Prefecture related to Research and Development in Radiological Sciences, the Agency for Natural Resources and Energy, the Ministry of Economy, Trade and Industry (METI), Japan, the JSPS KAKENHI (grant number JP17k00537, 17H01874), and partly supported by the Interdisciplinary Project on Environmental Transfer of Radionuclides. W. Men thanks the China Scholarship Council for financial support to carry out research abroad. The authors heartfully thank the editor as well as two anonymous reviewers for their constructive comments which helped to improve the manuscript.
Author information
Authors and Affiliations
Contributions
J.Z., W.M. and T.A. designed the study. W.M. performed the experiments on method development, data analysis and wrote the paper under the guidance of J.Z., S.L.M., K.T., S.U., M.Y. and T.A. and M.Y. collected seawater samples and provided important advices for the paper’s accomplishment. W.M., H.W. and Y.N. analyzed Pu isotopes in seawater samples collected in July 2013. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Men, W., Zheng, J., Wang, H. et al. Establishing rapid analysis of Pu isotopes in seawater to study the impact of Fukushima nuclear accident in the Northwest Pacific. Sci Rep 8, 1892 (2018). https://doi.org/10.1038/s41598-018-20151-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-20151-4
This article is cited by
-
A simple procedure for highly efficient purification of ultratrace Pu from Pb by extraction chromatography
Journal of Radioanalytical and Nuclear Chemistry (2022)
