
Abstract
A cell-free DNA (cfDNA) assay would be a promising approach to early cancer diagnosis, especially for patients with dense tissues. Consistent cfDNA signatures have been observed for many carcinogens. Recently, investigations of cfDNA as a reliable early detection bioassay have presented a powerful opportunity for detecting dense tissue screening complications early. We performed a prospective study to evaluate the potential of characterizing cfDNA as a central element in the early detection of dense tissue breast cancer (BC). Plasma samples were collected from 32 consenting subjects with dense tissue and positive mammograms, 20 with positive biopsies and 12 with negative biopsies. After screening and before biopsy, cfDNA was extracted, and whole-genome next-generation sequencing (NGS) was performed on all samples. Copy number alteration (CNA) and single nucleotide polymorphism (SNP)/insertion/deletion (Indel) analyses were performed to characterize cfDNA. In the positive-positive subjects (cases), a total of 5 CNAs overlapped with 5 previously reported BC-related oncogenes (KSR2, MAP2K4, MSI2, CANT1 and MSI2). In addition, 1 SNP was detected in KMT2C, a BC oncogene, and 9 others were detected in or near 10 genes (SERAC1, DAGLB, MACF1, NVL, FBXW4, FANK1, KCTD4, CAVIN1; ATP6V0A1 and ZBTB20-AS1) previously associated with non-BC cancers. For the positive–negative subjects (screening), 3 CNAs were detected in BC genes (ACVR2A, CUL3 and PIK3R1), and 5 SNPs were identified in 6 non-BC cancer genes (SNIP1, TBC1D10B, PANK1, PRKCA and RUNX2; SUPT3H). This study presents evidence of the potential of using cfDNA somatic variants as dense tissue BC biomarkers from a noninvasive liquid bioassay for early cancer detection.
Similar content being viewed by others

Introduction
Breast cancer (BC) is the most prevalent cancer worldwide, with an estimated 2.3 million new cases in 20201. According to the GLOBOCAN Cancer Tomorrow Prediction, incidences are expected to increase by 33.8% by 2040, suggesting a staggering 3 million new cases2. The incidence of mortality due to BC remains high in low-income countries due in part to the noticeable lack of options for early detection and therapy management3. In Tunisia, approximately 32.2 incident cases and 10.3 related deaths per 100.000 women were reported in late 20194. Currently, mammography is the only noninvasive method for detecting evidence of possible BC in dense tissue patients, and ultrasound-assisted core needle biopsy is the only robust and effective means of obtaining definitive diagnosis and staging of BC. Together, they provide a tenuous tandem method for accurately detecting early BC in dense tissue patients. Mammography has low sensitivity, with up to 34% false negative diagnoses for female dense tissue patients under 405,6. Complementary invasive ultrasound-assisted core needle biopsy has a number of shortcomings, including difficulty in targeting small lesions and the ability to miss underestimated lesions7. In addition, the mammography-tissue biopsy tandem does not provide detailed information (such as genetic mutations) that could be of great value in obtaining a precise diagnosis and delivering optimized therapy7. Collectively, these limitations suggest the untapped value of a more refined, robust, information-rich, noninvasive approach that reduces the need for repeated biopsies, unnecessary surgeries, and nonideally treatments, especially for women with dense breast tissue. In this context, liquid biopsy based on a simple noninvasive blood test is a very promising approach for investigating the tumor-derived material circulating in the bloodstream shed from primary tumors and their metastatic sites8. Among the tumor components in bodily fluids identified during the past decade, increasing attention has been given to circulating tumor DNA (ctDNA), which is now considered useful for the early detection and management of solid tumors such as those of colorectal, prostate and lung cancers9. The small nucleic acid fragments known as ctDNA (approximately 134–144 bp) are associated with abnormal cell structures and altered mechanisms10. Prior investigations have largely shown a high concordance between the ctDNA molecular profile and traditional tumor tissue using the same testing protocols11. Advances in next-generation sequencing (NGS) have simplified and improved the speed of the molecular identification and testing of ctDNA genomic alterations, proving value for novel target variant identification with the potential to improve patient outcomes12. Molecular investigations have demonstrated that the BC patient genome include somatic mutations and copy number alterations (CNAs) that correlate with cancer susceptibility and staging13. These genetic alterations can be detected in ctDNA from BC patients and thus are candidates for early BC detection and improved screening programs14. However, there are limited data regarding the variant profile differences among dense tissue subjects with positive mammograms and positive ultrasound biopsy versus those with positive mammograms and negative ultrasound biopsy against ctDNA molecular testing. In this study, we aimed to assess the differences in somatic variant profiles, including CNAs), single nucleotide polymorphisms (SNPs), and insertions/deletions (Indels), between subjects with positive mammograms and positive biopsies (pos-pos) versus subjects with positive mammograms and negative biopsies (pos-neg) using a ctDNA assay and to examine the differences in BC early detection and clinical outcomes of ctDNA testing.
Methods
Cohort
A cohort of 32 subjects with dense tissue and positive mammograms from Salah Azaiz Institute in Tunisia between June 2019 and January 2020 was recruited into the study. Clinical information was obtained through the medical records and a personal interview during sample collection. Cell-free DNA (cfDNA) sample collection was conducted after a positive mammogram but before ultrasound-assisted core needle biopsy. Microbiopsy test results were documented after confirmation by two independent physicians (radiologist and oncologist). This research was conducted through an Institutional Review Board-approved protocol (ISA/2019/04), and all subjects provided written informed consent for our study.
Sample preparation and cfDNA sequencing
Ten milliliters of peripheral blood samples were obtained immediately before ultrasound-guided core needle biopsy. Plasma from Streck BCT tubes was prepared within 2 h after blood collection and stored at − 20 °C in the clinic until shipment to the research laboratory. cfDNA was isolated from 5 ml of plasma with a MagMAX Cell-Free DNA Isolation Kit (MM; Applied Biosystems, Thermo Fisher Scientific, Foster City, CA, USA) and then eluted in 60 µl of elution buffer according to the manufacturer’s protocol. cfDNA was quantified using a QuantiFluor dsDNA System and GloMax Discover Microplate Reader (Promega, Madison, WI, USA). The distribution of fragment lengths was checked by electrophoresis on an Agilent 2100 Bioanalyzer with a High Sensitivity Large Fragment 50 kb DNA Kit (Agilent, Technologies Inc., Santa Clara, CA, USA). An NEBNext Ultra II DNA Library Prep kit (New England Biolabs, UK; E7645) was used for cfDNA whole-genome library preparation. Higher-pass whole-genome sequencing was started with 10 ng of cfDNA input (median of 5 ng). Finally, 32 libraries were pooled and sequenced using 150 bp pair-end run reads and 8 bp dual-indices on an Illumina NovaSeq machine (Illumina, San Diego, CA, USA), producing cfDNA whole-genome sequences for each subject.
Pathologic assessment and subject segregation
Pathologic tissues obtained by ultrasound-guided biopsy and under mammography for the whole cohort were reviewed by designated breast pathologists from Salah Azaiz Institute in Tunisia. According to the evaluation results from standard histology and mammogram imaging, the cohort was classified into two groups: the screening group, corresponding to subjects with positive mammography and negative biopsy (pos-neg; N = 12) and the cases group, corresponding to subjects with positive mammography and positive biopsy (pos-neg; N = 20). The absence of tumoral tissue as confirmed by examination was designated a “negative” biopsy, and a designation of a “positive” biopsy was made if the sample indicated stage I or II breast malignancy according to the 8th Edition of the American Joint Committee on Cancer (AJCC) Staging Manual for breast cancer15.
cfDNA sequence analysis
The analysis workflow performed in this study is summarized in Fig. 1. First, cfDNA whole-genome sequencing data were stored in Fastq files and then adapter trimmed using fastp (version 0.19.10) with default settings and -p-detect_adapter_for_pe16. The paired-end reads were aligned with BWA (version 0.7.17-r1188)17 to the GRCh38 human reference genome. The resulting BAM files were processed using the Picard (version 2.18.9) UmiAwareMarkDuplicatesWithMateCigar function (http://broadinstitute.github.io/picard/) to remove duplicate reads. FastQC (version 0.11.9) was run before and after adapter trimming to impose Fastq record quality control18, and Picard CollectWGSMetrics was used for BAM file quality control (http://broadinstitute.github.io/picard/).

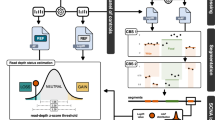
Schematic representation of the analysis workflow. cfDNA cell-free DNA, QC quality control, CNA copy number alterations, SNPs single nucleotide polymorphisms, Indels Insertion/deletions, CADD combined annotation dependent depletion, UCSC University of California Santa Cruz.
CNA
ichorCNA (version 0.3.2, https://github.com/broadinstitute/ichorCNA) was then applied to all high-quality aligned reads for each subject’s BAM files to estimate the tumor-derived DNA fraction (TF) and detect CNAs using all recommended default parameters except parameter adjustment to account for low cfDNA content samples19. Given the absence of an established control reference CNA set for these samples, no false-positive filtering was performed. Subsequently, the detected CNAs were grouped by subject status into “pos-pos” and “pos-neg” groups. The CNAs collected for each group were filtered to include only those shared by at least 2 subjects in the group and thereafter filtered to include alterations exclusive to that same group. These pos-pos and pos-neg exclusive CNAs were separately tested to determine the genes with which they overlapped using the UCSC Genome Browser20. The CNA-tagged genes were then tested against the Cancer Genes set found in the Precision Oncology Knowledge Base (OncoKB, 27) to determine which cancers (if any) the genes were associated with. These CNA-tagged cancer genes were then tested against the Candidate Cancer Gene Database21 to identify predicted associated cancers.
SNPs and indels
Grouped by pathology type (pos-pos; pos-neg), each subject’s BAM files were then analyzed by the Mutect2 part of GATK (v. 4.1.8.1)22 to detect somatic SNPs and Indels within the 22 autosomes against a ‘panel of normals’ created from the 1000 Genomes project23 and the gnomAD24 database as a ‘germline-resource’ included in the GATK resource bundle (https://console.cloud.google.com/storage/browser/genomics-public-data/resources/broad/hg38/v0). Identified variants were then filtered using GATK FilterMutectCalls22 using the recommended default parameters and thereafter annotated using ANNOVAR25. Variants with a minor allele frequency (MAF) > = 1% in the 1000 Genomes and ExAC databases were excluded26. Subsequently, candidate variants without a predicted deleterious nature were removed from consideration. To detect deleterious mutations, all variants were ranked using the CADD database (version 1.6), and those with a PHRED scaled score of > 10 were considered as having a probable deleterious function and retained in their respective pos-pos and pos-neg grouped collection27. For coding variants, the deleterious nature was predicted by MutationTaster28, PolyPhen V229, Provean30, and SIFT31, provided by the dbNSFP database (version 4.1)32. The grouped variants predicted to be deleterious by at least three of the four prediction engines were retained. For noncoding variants, the designation of ‘deleterious’ was assigned after application of SNPNexus33 and a threshold of FunSeq2 score > = 1.534. The coding and noncoding deleterious variants were then collected into the pos-pos and pos-neg groupings. As with the candidate cfDNA CNAs, candidate cfDNA SNPs and Indels were filtered to include those appearing in at least two individuals within the group and thereafter exclusive to either pos-pos or pos-neg groups. These pos-pos and pos-neg exclusive variants were then used to identify their associated genes and the subsequent determination of cancer association using the Candidate Cancer Gene Database21.
Statistical analysis
Statistical analysis was performed with R (version 3.6.2)35. Continuous variables are expressed as the means ± SDs, while categorical data are expressed as percentages of the total. Independent sample t tests were applied for intergroup comparisons of normally distributed continuous data, and chi-square tests were applied for categorical variables. P < 0.05 was considered statistically significant. The tumor fraction estimation boxplots of groups were created with the R-ggplot2 package36.
Ethical approval and consent to participate
All subject investigations conformed to the principles outlined in the Declaration of Helsinki and have been performed with permission of the study protocol approved by the ethics committee of Salah Azaiz Institute (SAI), under same’s Ethics Committee registration number (#ISA/2019/04). All subjects were informed about the purposes of the study and consented in writing to participate in the study.
Results
Cohort
A total of 32 women with dense breast tissue and a positive screening mammogram were recruited before microbiopsy. Detailed clinicopathological characteristics of the cohort are described in Table 1. Blood samples were acquired from all subjects for cfDNA analysis. Tumor status was confirmed by the pathology report from nodule biopsy and subsequent ultrasound. A cohort of 12 subjects with no confirmed tumors were stratified as pos-neg (age: 42.00 ± 4.73, BMI: 31.29 ± 6.53); 33.33% had a family history of nonbreast cancer. The remaining 20 subjects with confirmed tumors, 11 in stage I and 9 in stage II (age: 43.50 ± 3.95, BMI: 29.76 ± 5.07), were placed in the pos-pos group; 70% had a family history of nonbreast cancer, and 15% had a breast cancer history. No significant differences were observed between groups concerning the clinicopathological parameters (Table 1).
Tumor fraction estimation
The level of tumor‐derived DNA in plasma at baseline (after the positive mammogram and before microbiopsy) was predicted. Subjects were first analyzed as one group and then stratified based on the biopsy pathological results into four groups (pos-neg subjects and pos-pos Stage I, pos-pos Stage II and all pos-pos subjects). The lower limit of sensitivity for detecting the presence of tumor or TF cutoff was set to 3%, as suggested by the authors of the ichorCNA software. For the pos-neg cohort, the mean TF was 0.016 (range 0.012–0.021), and for the all pos-pos group, the mean TF was 0.018 (range 0.009–0.058). The difference in mean TF between the two groups was not statistically significant (p0 = 0.53). The pos-pos TF range was wider, suggesting a larger deviance between TFs in the pos-neg group than in the pos-pos group. The mean TF for the pos-pos Stage I group was 0.014 (range 0.009–0.020) versus 0.022 (range 0.013–0.058) for the pos-pos stage II group; the differences between these groups and the pos-neg group were not significant (p1 = 0.27 and p2 = 0.28, respectively). The mean TF differences between the pos-pos Stage I and II groups was also not statistically significant (p3 = 0.17), although the pos-pos Stage II group had a larger mean TF and contained the only subject with a TF above the 3% cutoff (Fig. 2).

Distribution of tumor fraction estimation. p0 Pos-neg vs. Pos-pos, p1 Pos-neg vs. Pos-pos Stage I, p2 Pos-neg vs. Pos-pos Stage II, p3 Pos-pos Stage I vs. Pos-pos Stage II. p-value: Student t-test, Pos-Neg Positive–negative subjects, Pos-Pos Positive-positive subjects.
CNAs and associated genes
CNA analysis detected a total of 1253 CNAs across all subjects, 1105 of which were in the pos-neg group and 868 in the pos-pos group. A total of 720 CNAs were shared by both groups, 385 found solely in the pos-neg group and 148 in the pos-neg group. The 1105 pos-neg CNAs were classified as gain (306), deletion (748) and amplification51. Of the 868 pos-pos CNAs, 382 were classified as gain, 435 as deletion and 51 as amplification (Fig. 3 and Table 2). Among the pos-neg subjects, chromosomes (Chr) 1 and 2 had the highest number of CNAs, 109 and 212, respectively, while for pos-pos cases, Chr 1 and 4 had 126 and 97 CNAs, respectively (Table 2). Of the 1253 total CNAs, 90 known overlapping oncogenes were identified; 15 were associated with CNAs found in both groups, 11 of which were previously described in cancers other than BC and 4 with a known association with BC. In addition, 49 deletion CNAs were detected in pos-neg subjects; 30 overlapped with genes previously described as associated with different cancers, 3 of which were previously associated with BC. On the other hand, 26 CNAs classified as gain were detected among the pos-pos subjects; 18 of these CNAs had a potential impact on genes that were previously described as associated with different cancers, 5 of which were described in BC (Table 3).

Diagram of the CNAs distribution in study groups. CNAs: Copy Number Alterations. *1: Exclusive CNAs detected in Pos-neg. *2: Shared CNAs between Pos-neg and Pos-pos. *3: Exclusive CNAs detected in Pos-pos. *4: Total CNAs for Pos-neg. *5: Total CNAs for Pos-pos. *6: Total CNAs detected in the study cohort (Pos-neg and Pos-pos). Pos-Neg: Positive–negative subjects; Pos-Pos: Positive-positive subjects.
SNPs, indels and associated genes
A total of 1,583,400 variants, 1,282,284 SNPs, 47,693 multiple nucleotide polymorphisms (MNPs) and 253,423 Indels were identified across all subjects before MAF and CADD filtering, which subsequently yielded 1,467,158 (1,215,768 SNPs, 47,693 MNPs and 203,697 Indels) and 143,719 variants, respectively (134,929 SNPs, 2386 MNPs and 6404 Indels). Of these 143,719 variants, 9494 and 134,225 were identified as coding and noncoding variants, respectively. Of the 9494 total coding variants, 3196 were predicted to have deleterious impact; out of these variants, 2139 were exclusive to the pos-pos group, and 1048 were exclusive to the pos-neg group. Subsequently, 10 variants were identified as shared by at least 2 subjects, 6 for the pos-pos group and 4 for the pos-neg group. Of the 134,225 noncoding variants detected, 78,704 were exclusive to the pos-pos group, and 38,845 were exclusive to the pos-neg group. Thereafter, 3992 and 1144 variants were identified as shared by at least 2 subjects of each group, respectively. Functional annotation of the noncoding variants identified 7 intronic variants, 5 in pos-pos and 2 in pos-neg subjects, and 3 upstream and downstream variants, 2 in pos-pos and 1 in pos-neg subjects (Table 4). A final set of 25 variants overlapped with oncogenes. Eighteen variants were identified among the pos-pos subjects (6 coding and 12 non-coding), and 10 of these 18 variants were previously described to be associated with liver, blood, pancreatic and skin cancers; only one pos-pos variant, rs2884935, was found in a gene (KMT2C) associated with BC. Among the pos-neg subjects, 7 variants were related to oncogenes (4 coding and 3 non-coding), and 5 of these were associated with blood, colorectal and pancreatic cancers, but none were detected in the breast oncogenes (Table 5).
Discussion
Multiple studies have demonstrated the significance of a noninvasive ctDNA variant testing biopsy for the early detection of solid tumors and subsequent improved outcomes37, therapy management38, response assessment39, and tumor resistance40. Short-fragment, low tumor-fraction cfDNA testing presents a challenge to early detection efforts, however. These fragments were largely investigated in clinical applications related to treatment prediction, relapse, and drug resistance41. Most previous studies focused on cfDNA levels as a predictive biomarker for therapeutic response in solid cancers42. Recently, a large-scale study based on cfDNA concentration showed that variation in the cfDNA level in plasma is not related to patient outcome and thus suggested that cfDNA concentration could not serve as a reliable biomarker for cancer management43. However, investigating cfDNA molecular profiles remains a viable opportunity for evaluating their relationship in detecting and characterizing the patient’s cancer status. In this study, we report a combined analysis of cfDNA whole-genome profiles between subjects with positive mammograms and biopsies versus subjects with positive mammograms and negative biopsies and suggest the possible role of these differences in the early detection of BC and subsequent clinical diagnosis, precision treatment protocols, and hopefully improved outcomes.
According to our assessment of previous research, our study is the first to examine and propose a full ctDNA analysis, including CNA and SNP/Indel detection and characterization, for identifying breast tumors in dense tissue subjects before mammogram identification. We assert that such an approach, when demonstrated to be robust, could serve as a precision oncology application in early BC detection.
In this study, the mean TF (0.016 and 0.018 for the pos-neg and pos-pos groups, respectively) was lower than the 3% recommended TF cutoff. The low TFs obtained in this study may be related to the low sensitivity in detecting the presence of ctDNA in our sequenced data19. However, the TF ranges were larger in the pos-pos group than in the pos-neg group and thus are possibly a different indicator of the presence of cancer than the TF alone. In addition, a higher TF was found in pos-pos stage II than in pos-pos stage I, suggesting that the ctDNA fraction increases as a function of tumor progression. These results support the interpretation that the isolated DNA fragments were ctDNA, an interpretation consistent with previous liquid biomarker studies investigating cfDNA as an early detection and prognosis biomarker in BC44. Other studies have demonstrated the reliability of ctDNA biomarkers for cancer therapeutic decision-making, evaluating patients’ resistance to treatment45,46, and tracking tumor progression during and after therapy47,48. The results of this study identified deletion and gain CNAs exclusively found in pos-neg subjects that overlapped across 11 known oncogenes. Three of these genes, JAK1, FUBP1, and RBM15, are all associated with liver, blood, colorectal and pancreatic cancers; three, TPR, CDC73 and PIK3C2B are all associated with blood and colorectal cancers; and five, JUN, NEGR1, VTCN1, DDR2 and PBX1, are associated with blood, liver, pancreatic, sarcoma and gastric cancer, respectively. In addition, among the pos-neg subjects, three exclusive deletion CNAs overlapped with the ACVR2A, CUL3 and PIK3R1 oncogenes, which are associated with BC. Among the pos-pos subjects, five exclusive gain CNAs overlapped with the KSR2, MAP2K4, MSI2, CANT1 and MSI2 oncogenes, all previously associated with BC (Table 3). Differences in the detected deletion and gain CNAs associated with pos-neg and pos-pos subjects may be related to epigenetic modifications and their impact on somatic alterations leading to oncogenesis and tumor growth49. The precise differences in nucleosome positioning between tumor and normal cells have been described as actively involved in the footprints of transcription factors associated with oncogenesis detectable in cfDNA fragments50. The nuclear architecture responsible for gene structure and expression has been correlated with cfDNA nucleosome occupancies, suggesting the potential for the early-stage detection of cancer cells51. Recently, these same nucleosome footprints identified cell types shedding cfDNA whose molecular profile suggested involvement in multiple pathological states, including cancer52. cfDNA profiling was also found to be informative of tumor localization and progression53. Differential release of cfDNA was also correlated with tumor heterogeneity among patients diagnosed with similar cancers and thus could be a promising biomarker of therapy management54. The collective evidence from the current and previous studies suggests that CNAs previously described in breast tissue coupled to their presence in a ctDNA-based biopsy may play an important role in the early detection and diagnosis of BC. The SNP and Indel results identified 10 functionally important variants in the pos-pos subjects previously associated with cancer. One variant, rs757825963, was located in SERAC1, a known BC risk factor. In addition, SERAC1 is also associated with leukopenia55, and increased expression of SERAC1 has been correlated with BC risk56. SERAC1 also has a strong interaction with multiple splicing factors (hnRNP A3, hnRNP J, hnRNP G, FMRP, Fox-2) in the context of cancer prognosis and development57. The clear and important role of SERAC1 in splicing events suggests a likely role as an early detection liquid biopsy biomarker when coupled to the role of cfDNA variants associated with dysregulation related to epigenetics. Another identified variant, rs147494591, found in FBXW4, which encodes for the F-box proteins that are involved in biological processes such as cell growth, division, development, differentiation, survival and death58, suggests another possible molecular biomarker for early BC detection. Previous studies found that decreased expression of FBXW4 was correlated with poor survival among non-small-cell lung cancer patients59. A recent study showed that downregulation of FBXW4 favored colorectal tumor relapse and limited the survival range60. Together with the results of this study, these previous study findings suggest that FBXW4 may be an important prognostic indicator in oncology. Pos-pos subject variants identified in NVL suggest a role in the dysregulation of telomere function, possibly initiating breast tumor development. The depletion role of NVL was strongly associated with lower hTERT, associated with decreased telomerase activity in multiple pathogeneses61. Two exclusively pos-pos variants found in known BC risk-associated genes (FANK1 and KCTD4) suggest further pos-pos cfDNA somatic association with BC risk. FANK1 was recently identified as a novel binding partner in mammalian cells that prevents the proteasome degradation of polyubiquitinated FANK1, which leads to the activation of the AP-1 signaling pathway and the induction of tumor cell apoptosis62. KCTD4 was reported as a tumor suppressor gene associated with insertional mutagenesis for leukemia or lymphoma development in insertional mutagenesis in a mouse model study63. The deregulation of both FANK1 and KCTD4 may be a consequence of the observed somatic variants, thus suggesting another association with tumor development and their use as an early detection biomarker in a cfDNA-based assay. The two pos-pos–associated variants (rs766835420 and rs190711126), located in DAGLB and CAVIN1/ATP6V0A1, respectively, were positively associated with BC. SNPs of DAGLB have been correlated with increased DAGLB expression in stomach tissues and were also significantly elevated in gastric tumors compared to adjacent tissues, thus confirming the potential of DAGLB as a susceptibility gene for gastric cancer64. Loss of stromal CAVIN1 expression negates the ability of stromal cells to sequester lipids and is associated with the upregulation of inflammatory factors such as cytokines and their receptors, matrix metalloproteinases, and markers for CAFs65. Deregulation of any inflammatory microenvironment factors, such as those seen in CAVINI, promotes aggressive cancer phenotypes, thus supporting the critical function of CAVINI in the stromal component in tumorigenesis and suggesting a metastasis-suppressing role for this gene66. Any deleterious variant appearing in CAVIN1 will likely contribute to lower CAVINI expression and loss of stromal cell function, suggesting a role in breast cancer genesis and tumor development. Other deleterious pos-pos variants found in MACF1 and ZBTB20-AS1 align with earlier studies showing that MACF1 mutations detected in tissue-specific genomes are responsible for function dysregulation associated with cancer67, and a correlation study found that key ZBTB20-AS1 lncRNAs are associated with colon tumor staging and likely tumor progression68. Finally, a pos-pos exclusive variant was associated with KMT2C, a known BC risk factor. In addition, KMT2C is the gene with the highest mutation count predominantly found in BC, with some mutations associated with chromatin function, affecting transcription mechanisms identified in breast tumor development69. KMT2C mutations were also shown to be key to ERα regulation, which can lead to hormone-driven breast cancer cell proliferation70. In summary, the somatic variants found in the pos-pos cases investigated in this study present a rich and highly associated set of potential biomarkers shown to affect key molecular mechanisms important to oncogenesis (and its suppression) and therefore may be putative biomarkers for early BC detection.
Concerning the pos-neg screening group, 6 oncogenes were identified as containing exclusive variants: SNIP1, TBC1D10B, PRKCA, RUNX2 and SUPT3H. PRKCA has been previously identified as associated with BC and encodes a calcium-dependent protein kinase involved in multiple biological functions, including calcium ion transport, exocytosis, cell growth, and proliferation71. PRKCA is also a central signaling node and coinhibitor of the ESR1, mTORC1, and HDAC genes known to suppress breast cancer72. The collective evidence suggests that PRKCA is an important candidate for breast carcinoma stem cell management73. Two hypotheses suggest a role for PRKCA somatic variants in the absence of cancer in pos-neg subjects. First, these variants may have a protective effect against BC oncogenesis via the modulation of PRKCA expression, thus delaying if not stopping tumor development and growth.
Despite the notable results, there are limitations to be acknowledged. This is a small subject study, and a large cohort study must follow to validate these results and thereby challenge the robustness of the proposed biomarkers. Additionally, it is important that an additional study be performed with healthy control subjects (neg-neg) to test for any BC-associated cfDNA variants. These studies should also include normal tissue (from all subjects) and tumor tissue samples (from pos-pos cases) to validate the cfDNA profile against the tumor profile, thus confirming that cfDNA is actually ctDNA. TF levels must also be tested against presence and staging to further validate the use of TF range and low TF to confirm tumor presence and absence. Some detected variants in the pos-pos case group were previously detected in non-BC tumors. This result raises the possibility that such ctDNA variations may be present due to genome disorder, suggesting that these may not be valid biomarkers for BC.
Conclusions
Early breast cancer detection is of paramount importance in managing the most common cancer worldwide. Any bioassay suggested to be a robust test of early BC must be precise, repeatable, inexpensive and preferably noninvasive to replace the standard mammogram-biopsy protocol for BC diagnosis, but at this time, no such bioassay exists. Studies such as this in dense tissue subjects demonstrate promising evidence that a low-TF (thus providing early detection), noninvasive, robust bioassay may be available through cfDNA molecular testing. The presented results and suggestion are the first to describe a coupled analysis of CNA and SNP/Indel identification using cfDNA profiles for breast cancer early detection. Before these promising results can be used in the development of a panel of biomarkers for a biopsy, further understanding of early breast tumor biology and of the mechanisms that lead to tumor progression, is greatly needed to identify the molecular biomarkers to be used with such a highly informative assay. The molecular profiling and analysis workflow performed in this study on cfDNA taken from early screened and confirmed BC subjects presents promising results contributing to the knowledge required to create such a liquid biopsy test. Further investigations building on this are needed to confirm the results of this study, test the putative cfDNA molecular biomarkers and confirm their validity for inclusion in an early BC detection bioassay. In this way, these biomarkers could can contribute to significant improvements in BC diagnosis and therefore improved treatment optimization and subsequent outcomes to reduce the devastating incidence and mortality of breast cancer.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- cfDNA:
-
Cell-free DNA
- BC:
-
Breast cancer
- CNAs:
-
Copy number alterations
- ctDNA:
-
Circulating tumor DNA
- NGS:
-
Next-generation sequencing
- SNPs:
-
Single nucleotide polymorphisms
- Indels:
-
Insertions/deletions
- MM:
-
MagMAX
- TF:
-
Tumor fraction
- MAF:
-
Minor allele frequency
- MNPs:
-
Multiple nucleotide polymorphisms
References
20-Breast-fact-sheet.pdf.
Cancer Tomorrow [Internet]. [cited 2021 Feb 5]. Available from: https://gco.iarc.fr/tomorrow/en
Lei, S. et al. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 41, 1183–1194 (2021).
Kahale LA, Ouertatani H, Brahem AB, Grati H, Hamouda MB, Saz-Parkinson Z, et al. Contextual differences considered in the Tunisian ADOLOPMENT of the European Guidelines on Breast Cancer Screening [Internet]. In Review; 2020 Sep [cited 2021 Feb 5]. Available from: https://www.researchsquare.com/article/rs-72256/v1
Wang, L. Early diagnosis of breast cancer. Sensors 17, 1572 (2017).
Identification and validation of plasma biomarkers for diagnosis of breast cancer in South Asian women | Scientific Reports. https://www.nature.com/articles/s41598-021-04176-w.
Peled, M. et al. Cell-free DNA concentration in patients with clinical or mammographic suspicion of breast cancer. Sci. Rep. 10(1), 14601 (2020).
Tzanikou, E. & Lianidou, E. The potential of ctDNA analysis in breast cancer. Crit. Rev. Clin. Lab. Sci. 57(1), 54–72 (2020).
Song, Q., Zhang, Y., Liu, H. & Du, Y. Potential of using cell-free DNA and miRNA in breast milk to screen early breast cancer. Biomed. Res. Int. 3(2020), 1–11 (2020).
Stewart, C. M. et al. The value of cell-free DNA for molecular pathology. J. Pathol. 244(5), 616–627 (2018).
Toor, O. M. et al. Correlation of somatic genomic alterations between tissue genomics and ctDNA employing next-generation sequencing: Analysis of lung and gastrointestinal cancers. Mol. Cancer Ther. 17(5), 1123–1132 (2018).
Horak, P., Fröhling, S. & Glimm, H. Integrating next-generation sequencing into clinical oncology: Strategies, promises and pitfalls. ESMO Open. 1(5), e000094 (2016).
Azim, H. A., Nguyen, B., Brohée, S., Zoppoli, G. & Sotiriou, C. Genomic aberrations in young and elderly breast cancer patients. BMC Med. 13(1), 266 (2015).
Clifton, K. et al. Mutation profile differences in younger and older patients with advanced breast cancer using circulating tumor DNA (ctDNA). Breast Cancer Res Treat. https://doi.org/10.1007/s10549-020-06019-0 (2020).
Giuliano, A. E., Edge, S. B. & Hortobagyi, G. N. Eighth edition of the AJCC cancer staging manual: Breast cancer. Ann. Surg. Oncol. 25(7), 1783–1785 (2018).
fastp: an ultra-fast all-in-one FASTQ preprocessor | Bioinformatics | Oxford Academic. https://academic.oup.com/bioinformatics/article/34/17/i884/5093234?login=true.
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14), 1754–1760 (2009).
Home - BioBam - Bioinformatics Made Easy. BioBam https://www.biobam.com/.
Adalsteinsson, V. A. et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 8, 1324 (2017).
Navarro Gonzalez, J. et al. The UCSC genome browser database: 2021 update. Nucl. Acids Res. 49, 1046–1057 (2021).
Abbott, K. L. et al. The candidate cancer gene database: A database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res. 43, 844–848 (2015).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Auton, A. et al. A global reference for human genetic variation. Nature 526(7571), 68–74 (2015).
The mutational constraint spectrum quantified from variation in 141,456 humans | Nature. https://www.nature.com/articles/s41586-020-2308-7.
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38(16), e164–e164 (2010).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46(3), 310–315 (2014).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 7(8), 575–576 (2010).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods. 7(4), 248–249 (2010).
Choi, Y. & Chan, A. P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31(16), 2745–2747 (2015).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31(13), 3812–3814 (2003).
Liu, X., Li, C., Mou, C., Dong, Y. & Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12(1), 103 (2020).
Oscanoa, J. et al. SNPnexus: A web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 48, W185–W192 (2020).
Fu, Y. et al. FunSeq2: A framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 15(10), 480 (2014).
Bunn, A., Korpela, M. Crossdating in dplR. :12.
Villanueva, R. A. M. & Chen, Z. J. ggplot2: Elegant graphics for data analysis. Meas. Interdiscip. Res. Perspect. 17(3), 160–167 (2019).
Chera, B. S. et al. Plasma circulating tumor HPV DNA for the surveillance of cancer recurrence in HPV-associated oropharyngeal cancer. J. Clin. Oncol. 38(10), 1050–1058 (2020).
Tie, J. et al. Circulating tumor DNA analyses as markers of recurrence risk and benefit of adjuvant therapy for stage III colon cancer. JAMA Oncol. 5(12), 1710–1717 (2019).
Christensen, E. et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. JCO. 37(18), 1547–1557 (2019).
Horn, L. et al. Monitoring therapeutic response and resistance: Analysis of circulating tumor DNA in patients with ALK+ lung cancer. J. Thorac. Oncol. 14(11), 1901–1911 (2019).
Kilgour, E., Rothwell, D. G., Brady, G. & Dive, C. Liquid biopsy-based biomarkers of treatment response and resistance. Cancer Cell 37(4), 485–495 (2020).
Kumar, S. et al. Plasma DNA level in predicting therapeutic efficacy in advanced nonsmall cell lung cancer. Eur. Respir. J. 36(4), 885–892 (2010).
Pan, S. et al. Can plasma DNA monitoring be employed in personalized chemotherapy for patients with advanced lung cancer?. Biomed. Pharmacother. 66(2), 131–137 (2012).
Li, B. T. et al. A prospective study of total plasma cell-free DNA as a predictive biomarker for response to systemic therapy in patients with advanced non-small-cell lung cancers†. Ann. Oncol. 27(1), 154–159 (2016).
Fernandez-Garcia, D. et al. Plasma cell-free DNA (cfDNA) as a predictive and prognostic marker in patients with metastatic breast cancer. Breast Cancer Res. 21(1), 149 (2019).
Choudhury, A.D., Werner, L., Francini, E., Wei, X.X., Ha, G., Freeman, S.S., et al. Tumor fraction in cell-free DNA as a biomarker in prostate cancer. JCI Insight [Internet]. [cited 2021 Feb 5];3(21). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6238737/
Housman, G. et al. Drug resistance in cancer: An overview. Cancers 6(3), 1769–1792 (2014).
Ng, S. B. et al. Individualised multiplexed circulating tumour DNA assays for monitoring of tumour presence in patients after colorectal cancer surgery. Sci. Rep. 7(1), 40737 (2017).
Conteduca, V. et al. Plasma tumour DNA as an early indicator of treatment response in metastatic castration-resistant prostate cancer. Br. J. Cancer 123(6), 982–987 (2020).
Sun, K. et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. PNAS 112(40), E5503–E5512 (2015).
Kang, H., Hata, A. Chapter six-control of drosha-mediated microRNA maturation by smad proteins. In: Guo F, Tamanoi F, editors. The Enzymes [Internet]. Academic Press; 2012 [cited 2021 Feb 5]. p. 123–36. (Eukaryotic RNases and their Partners in RNA Degradation and Biogenesis, Part B; vol. 32). Available from: https://www.sciencedirect.com/science/article/pii/B9780124047419000064
Chromatin - an overview | ScienceDirect Topics [Internet]. [cited 2021 Feb 5]. Available from: https://www.sciencedirect.com/topics/neuroscience/chromatin
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M. & Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164(1), 57–68 (2016).
Gundem, G. et al. The evolutionary history of lethal metastatic prostate cancer. Nature 520(7547), 353–357 (2015).
Brastianos, P. K. et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 5(11), 1164–1177 (2015).
Svedberg, A. et al. Genetic association of gemcitabine/carboplatin-induced leukopenia and neutropenia in non-small cell lung cancer patients using whole-exome sequencing. Lung Cancer 1(147), 106–114 (2020).
Kar, S. P. et al. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 6(9), 1052–1067 (2016).
Zheng, Y., Shen, Z., Fan, Z., Wang, W., Geng, Q., Kan, Q., et al. Alternative splicing events and subtype analysis of esophageal cancer [Internet]. In Review; 2020 Oct [cited 2021 Feb 5]. Available from: https://www.researchsquare.com/article/rs-80935/v1
Skaar, J. R., Pagan, J. K. & Pagano, M. Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 14(6), 369–381 (2013).
Lockwood, W. W., Chandel, S. K., Stewart, G. L., Erdjument-Bromage, H. & Beverly, L. J. The novel ubiquitin ligase complex, SCFFbxw4, interacts with the COP9 signalosome in an F-box dependent manner, is mutated, lost and under-expressed in human cancers. PLoS ONE 8(5), e63610 (2013).
Zhang, Y. et al. FBXW4 acts as a protector of FOLFOX-based chemotherapy in metastatic colorectal cancer identified by co-expression network analysis. Front. Genet. https://doi.org/10.3389/fgene.2020.00113/full?report=reader (2020).
Wang, M. et al. The NVL gene confers risk for both major depressive disorder and schizophrenia in the Han Chinese population. Prog. Neuropsychopharmacol. Biol. Psychiatry 1(62), 7–13 (2015).
Ma, W. et al. Proapoptotic RYBP interacts with FANK1 and induces tumor cell apoptosis through the AP-1 signaling pathway. Cell. Signal. 28(8), 779–787 (2016).
Jofra Hernández, R. et al. Hematopoietic tumors in a mouse model of X-linked chronic granulomatous disease after lentiviral vector-mediated gene therapy. Mol. Ther. 29(1), 86–102 (2021).
Ni, J. et al. Integration of GWAS and eQTL analysis to identify risk loci and susceptibility genes for gastric cancer. Front Genet. https://doi.org/10.3389/fgene.2020.00679/full?report=reader (2020).
Low, J.-Y. et al. Stromal CAVIN1 controls prostate cancer microenvironment and metastasis by modulating lipid distribution and inflammatory signaling. Mol. Cancer Res. 18(9), 1414–1426 (2020).
Full article: Single nucleotide polymorphism mutation related genes in bladder cancer for the treatment of patients: a study based on the TCGA database [Internet]. [cited 2021 Feb 5]. Available from: https://www.tandfonline.com/doi/full/https://doi.org/10.1080/13102818.2020.1864231
Qian, W. et al. Construction of ceRNA networks reveals differences between distal and proximal colon cancers. Oncol. Rep. 41(5), 3027–3040 (2019).
Argyri, M., Viktor, L., Malin, M., Arendt, M.L., Jessika, N. Link to external site this link will open in a new window, et al. Targeted sequencing reveals the somatic mutation landscape in a Swedish breast cancer cohort. Scientific Reports (Nature Publisher Group) [Internet]. 2020 [cited 2021 Feb 5];10(1). Available from: https://search.proquest.com/docview/2471554712/abstract/45F07C679DAC4029PQ/1
Gala, K. et al. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERα enhancer function. Oncogene 37(34), 4692–4710 (2018).
Chen, J. et al. Identification of key candidate genes involved in melanoma metastasis. Mol. Med. Rep. 20(2), 903–914 (2019).
Sulaiman, A. et al. Co-inhibition of mTORC1, HDAC and ESR1α retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis. 9(8), 1–14 (2018).
Zhang, Z., Chen, X., Zhang, J. & Dai, X. Cancer stem cell transcriptome landscape reveals biomarkers driving breast carcinoma heterogeneity. Breast Cancer Res. Treat. https://doi.org/10.1007/s10549-020-06045-y (2021).
Acknowledgements
We thank all blood donors who participated in the present study. We express our thanks to Drs. Eduardo J. Simoes and Balkiss Bouhaouala-Zahar for their excellent assistance with experiments, discussion of results and suggested ideas for consideration.
Funding
This work was supported in by funding provided by the Center for Biomedical Informatics, School of Medicine, University of Missouri, Columbia.
Author information
Authors and Affiliations
Contributions
M.B.: Participated in study design, carried out the study and managed all project study participants who aided with experiments, patient consenting and chart, data review, manuscript preparation and data analysis. A.A.M.: Data analysis and processing, variant calling, and manuscript editing. E.G.: Participated in study design, data processing, sequencing alignment and editing manuscript. A.M.: Clinical data acquisition update and review. A.Z.: Patient recruitment and patients pathological report confirmation. N.B.: discussion of results and review of manuscript. P.J.T.: Project principal investigation, original idea, study concept and design, guided overall study analysis, discussion of results, supervised the bioinformatics and statistical data analysis and interpretation, review of manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Erik Gafni and Nathan Boley are employees of Ravel Biotechnology Startup. The remaining authors have no conflict.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barbirou, M., Miller, A.A., Gafni, E. et al. Evaluation of cfDNA as an early detection assay for dense tissue breast cancer. Sci Rep 12, 8458 (2022). https://doi.org/10.1038/s41598-022-12457-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-022-12457-1
This article is cited by
-
Enhancer regulatory networks globally connect non-coding breast cancer loci to cancer genes
Genome Biology (2025)
-
Elucidating the role of FBXW4 in osteoporosis: integrating bioinformatics and machine learning for advanced insight
BMC Pharmacology and Toxicology (2025)
-
Genomic landscapes of breast cancer in African populations: a systematic review
npj Breast Cancer (2025)
-
Use of ctDNA in early breast cancer: analytical validity and clinical potential
npj Breast Cancer (2024)
