
Abstract
Non-precious electrocatalysts as the alternative to Pt have become a hot research area in the last decade due to the suitable catalytic activity in Oxygen reduction reaction (ORR) in electrochemical systems. In this work, the density functional theory calculations were investigated to explore the activity of Fe, Cu, and Fe-Cu atoms supported by N-doped graphene as the ORR electrocatalyst for Oxygen-depolarized cathodes (ODCs). To this end, the ORR mechanism was surveyed in detail in the gas and solvent phases. The results show that the solvent phase leads to a higher overpotential and thermodynamic limiting potential. According to the density of states curves, there are strong interactions between metal atom and substrate that can effectively tune the electronics of catalysts. Bader's analysis confirms that, in addition to the single metal atoms, nitrogen atoms have also played a critical role in charge transfer between substrates and oxygen molecules in ORR. It is also predicted that Fe-Cu@NC SAC exhibits the highest catalytic activity which is consistent with thermodynamic limiting potential and theoretical overpotential of − 0.26 and 0.66 (V vs. SHE), respectively, indicating that this type of catalyst may be a suitable candidate instead of precious metals in oxygen-depolarized cathodes in electrochemical devices.
Similar content being viewed by others
Introduction
Oxygen-depolarized cathodes (ODCs) are indispensable components within a variety of electrochemical systems, encompassing metal-air batteries, fuel cells, and electrolyzers1,2,3,4. The Oxygen Reduction Reaction (ORR) occurring at ODCs serves as a cornerstone for effective energy conversion and storage processes. Understanding the intricacies and nuances of ORR in ODCs is paramount for elevating device performance, longevity, and overall efficiency5. Moreover, ORR plays a pivotal role in oxygen-depolarized electrolyzers, where it drives the conversion of oxygen into hydroxide ions using electrical energy. Addressing the challenges associated with ORR in ODCs demands interdisciplinary collaboration, uniting expertise from fields such as materials science, electrochemistry, and device engineering6,7. Through the integration of recent advancements and the exploration of novel research avenues, the development of efficient, cost-effective, and sustainable ORR-based ODCs holds tremendous potential for propelling the realm of electrochemical energy conversion forward, thus contributing to a cleaner and more sustainable energy landscape8,9,10,11,12,13.
ORR has sluggish kinetics in both acidic and alkaline media, and the recent research focused on ORR electrocatalysts, including platinum group metal (PGM), non-PGM, carbon-based materials, and single-atom catalysts14,15,16,17,18,19. Alongside experimental works, the computational approaches were frequently applied to evaluate the ORR on platinum-based catalysts and other metals20,21,22,23,24,25. Nørskov et al.26 explained that Pt is the best metal catalyst among different transition and noble metals for ORR in acidic media by a volcano relationship between the rate of the cathode reaction and the intermediates binding energy (*O or *OH). Zhang et al.27 systematically studied the ORR at various Pt–Bi surfaces by DFT calculations. Their results showed that the introduction of Bi into Pt changes the potential-determining step (RDS) of ORR and PtBi (100) structure, with the lowest d-band center, gives the best ORR activity compared to the Pt (111). Gao28 explored the mechanism of the ORR on M–N3 (M = Mn, Co, Ni) co-doped defective graphene. At pH = 0, the overpotentials of ORR for Mn-N3-Gra, Co-N3-Gra, and Ni-N3-Gra are 0.56 V, 0.849 V, and 0.381 V, respectively. These results indicated that Ni-N3-Gra is comparable with Pt. It is also to be mentioned that most of the computational investigations of the ORR have concentrated on acid solutions and there are few theoretical studies have been conducted on alkaline conditions. As an example of studying in an alkaline environment, we can refer to Yu et al.29. They have studied two ORR mechanisms, i.e., dissociative and associative mechanisms, over different N-doped graphene on the cathode of fuel cells by DFT calculations. In this study, it can be seen that the ORR path through the associative mechanism is more favorable than the dissociative one by comparing the free energy curves. The results indicated the rate-determining step is the removal of *O from the surface of the N-doped configuration. Their results illustrated that this ORR catalyst is promising to replace Pt in the alkaline medium. Ignaczak et al.30 proposed a complete reaction sequence of ORR in alkaline solutions by comparing the adsorption of O2 on three metals such as Au(100), Ag(100), and Pt(100). According to the first step in ORR (O2 + e− → \({O}_{2}^{-}\)), they claimed that a small adsorbate cannot exist in two diverse charge states on a surface. Likewise, the reaction rate on a metal surface cannot be affected by the electrode potential. So at least one of the reactants must be in the outer sphere. Due to the strong electronegativity of oxygen, it is adsorbed in the form of the \({O}_{2}^{-}\) on Ag (100), and both the inner sphere and the outer sphere mechanism can occur in parallel. About Au (100), the first step of ORR takes place in the outer sphere mode, and on Pt (100), the adsorbed state is favorable. They have investigated that either reaction follows a 4e− or 2e− mechanism, depending on the adsorption energy of OH.
In order to properly interpret experimental results and predict the properties of new materials before synthesis, a detailed analysis by DFT calculations is needed. In our previous study, we conducted an in-depth analysis of the oxygen reduction reaction mechanism in alkaline media by DFT calculations, with a particular emphasis on single atoms of non-precious metals31. Through examination of Gibbs free energy profiles and the electrochemical-step symmetry index (ESSI), it was revealed that the SAC containing copper exhibited the most remarkable catalytic activity. This study is aimed to explore the ORR mechanism on single metal (Fe and Cu) and dual metal atom catalysts (Fe–Cu) supported on N-doped graphene considering solvent such as water (ε = 78.54) and the gas phase (ε = 1). This theoretical research in the gas and liquid phases can be a critical step towards understanding the complexity of cathodic reaction in Electrochemical devices and can be used as a guide for further studies.
ORR mechanism in alkaline media
The focus of this study is on the associative mechanism of ORR under alkaline media where O2 proceeds to OH− on the surface through four reaction steps as follows:
which involves the *O, *OH, and *OOH intermediates. We followed the mechanism from Liang et al.32 to compute Gibbs free energy change (ΔGi, i = 1–4) for each step and predict the theoretical overpotential for both gas and solvent phases. ΔGi values are calculated as follows:
According to the literature33,34, G* is the total energy of the clean catalyst model, so G(*) = E*, whereas for the intermediates (X = *OOH, *O, *OH) on the surface, we have:
where E(X) represents the total energy of the model with the adsorbed species X, EZPE(X) is the vibrational zero-point energy, and S(X) stands for the entropy of the adsorbed species. To calculate the only remaining terms G(OH−) − G(e−), we follow Liang et al. method which used the equilibrium as H2O(l) → H+ + OH−, for which it holds that G(OH−) + G(H+) = G(H2O(l)), adding and subtracting G(e−) in the left-hand side one leads to32
Substituting Eq. (10) into Eqs. (5)–(8), one gets
In addition, applying an electrochemical-thermodynamic approach35, we can introduce the potential relative to the SHE and the pH in Eqs. (11)–(14), then
where v(H+) and v(e−) are the numbers of transferred protons and electrons, kB is the Boltzmann constant, e is the elementary charge of an electron, and U is the applied electrode potential with respect to the SHE36. It is also necessary that we consider thermodynamic limiting potential (UL) for a real catalyst. UL is the highest potential at which all reaction steps are exergonic37 and is defined as
The theoretical overpotential (ƞth) serves as an indicator of the effectiveness of a catalyst activity, which is defined as the difference between the equilibrium potential (U0) and the limiting potential (UL), this is
U0 for ORR in alkaline media is equal to + 0.4 V vs. SHE. It is also important to note that for the ideal catalyst, all steps have the same ΔGi, and UL = U0 leading to ƞth = 0.
Computational details
Methods
All our calculations, in DFT framework, were carried out in Dmol3 code embedded in the Material studio38,39. To describe the exchange–correlation function, the generalized gradient approximation (GGA) with Perdew-Burke-and-Ernzerhof (PBE) function was used40. DFT Semi-core Pseudopotentials (DSPP) were employed to achieve valid results, and double numerical plus polarization (DNP) basis sets were chosen41. For describing the van der Waals type long-range forces between absorbent species and host surface, the approach of Grimme was used42. For the unit cell of 5 × 5 × 1 with lattice parameters (a = b = 12.3 Å, c = 20 Å), a 4 × 4 × 1 Monkhorst–Pack k-point sampling was utilized. The convergence tolerances of energy and smearing in the geometric optimization process were 1 × 10−5 Ha and 0.005 Ha, respectively. The conductor-like screening model (COSMO) is used to simulate the aqueous environment, where the dielectric constant was set as 78.54 (H2O)43.
Models
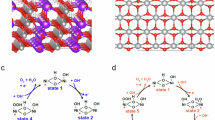
We consider a 5 × 5 × 1 supercell for graphene where two single metal atoms (Fe and Cu) are coordinated to six N atoms as indicated in Fig. 1. In this way, the metal atoms form two six- and one five-membered rings around them and one four-membered ring together. The final unit cell contains 40 C atoms, 6 N atoms, and 2 single metal atoms. To prevent interaction between periodic replicas, in periodic calculations, the 20 Å value is chosen as the third lattice parameter (c). In this study, the results are compared with the electrode model which includes one single atom (Cu or Fe) in the structure of a graphene supercell and four N atoms. The metal atom forms two five- and two six-membered rings (Fig. 1).

Schematic representation of the model of single and dual atom electrocatalysts in the present work. Grey, purple, orange, and light blue balls denote C, N, Cu, and Fe, respectively.
Results and discussion
Adsorption energy and electronic properties
Adsorption energy is a measure of how strongly an adsorbate binds to the surface of a catalyst. DFT calculations can provide valid information about the electronic and geometric properties of adsorbates (*O2, *OOH, *O, *OH) and substrates. First, we need to optimize the model and do the DFT calculations for three different cases: (1) the isolated surface of catalyst in the supercell, (2) the isolated adsorbate molecules, and (3) the combined surface + adsorbate system.
The DFT calculations will give us the total energies of each case, which we can use to calculate the adsorption energy using this formula:
where Esystem is the total energy of the optimized system, ESurface is the total energy of the bare surface and EX is the total energy of an isolated X adsorbate (X = *O2, *OOH, *O, *OH). A more negative the Eads illustrates a better thermodynamically favorable adsorption process44. Table 1 summarizes the obtained values including the adsorption energy of different optimized structures.
In the model of the single-atom catalyst, a single metal (Fe or Cu) is coordinated by four nitrogen atoms in the graphene plane. The average distances between the metal and nitrogen atoms are 1.89 Å and 1.92 Å for the Fe@NC and Cu@NC structures, respectively. Due to the presence of a vacuum defect, the bond lengths of all of M–N are longer than the C–C (1.42 Å) in pristine graphene, consistent with previously reported results45,46,47. Moreover, the average bond length of M–N in this study is smaller than the reported M–N bond length in the M-N3-doped graphene, with a range of 1.93–2.45 Å28. These results indicate that the covalent bonds between the single metal atom and the nitrogen atoms are stronger than the reported structure of Ref.28. For dual atoms catalyst (Fe-Cu@NC), the average bond length of Fe–N and Cu–N are 1.70 Å and 1.78 Å, respectively. Consequently, the bond strength in the structure of Fe-Cu@NC is stronger than Fe@NC and Cu@NC.
The electronic character is a critical parameter that directly affects the electrochemical performance of a catalyst. Therefore, we investigated this parameter by density of states (DOS) and projected density of states (PDOS) plots. The results illustrated the metallicity of our structures (Fig. S1). According to these results, there are overlapped electronic states (M3d-N2p: M = Fe and Cu) due to the strong covalent bonding between single metal atoms and four nitrogen atoms. The asymmetric DOS profiles at the Fermi level (zero) and around it, indicate a strong magnetic moments for our catalytic models. Moreover, the strong interaction between single atom-substrate can effectively tune the electronics of SAC. In addition, The d-band centers of Fe@NC, Cu@NC, and Fe-Cu@NC were −3.83, −4.19, and −2.39 eV, respectively (Fermi level = 0). Based on the PDOS analyses and d-band theory, an upshift of the d-band center for the electrocatalysts is attributed to the charge transfer between the metal sites. The d-band of Fe-Cu@NC shifts close to Fermi level, and enhances the capture of the O2 molecule that is the initial step of ORR47. The band gap refers to the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) in a material. Therefore, a catalyst with a smaller band gap may be more effective for reactions that involve electron transfer steps. The band structure graphs in Fig. S2 show the band gaps of Fe@NC, Cu@NC, and Fe-Cu@NC are 0.074, 0.447, and 0.033 eV, respectively. However, the choice of catalyst should not be solely based on the band gap. Other factors such as the specific reaction mechanism, the nature of reactants and intermediates, surface properties, and catalyst stability should also be considered.
Electron transferring has a key role in the performance of ORR electrocatalysis and the adsorption of O2. In this study, the useful Bader's theory of atoms in molecules is used for analyzing the charge. The assumption of maximum charge density in atomic centers (or at pseudoatoms) is considered in Bader's analysis48,49. In the ORR, firstly oxygen molecules adsorbed to the catalyst and gain electrons. In contrast, single metal atoms are the electron donor in this process. The average charge transfer for Fe and Cu are 1.08 and 0.90, respectively. This confirms that single metal atoms are suitable active sites that lead to considerable differences in charge transfer between substrates and oxygen molecules. The average charge transfers for C and N in all catalytic models are 0.09 and − 1.18, respectively. This confirms that, in addition to the single metal atoms, nitrogen atoms have also played an effective role in charge transfer in the oxygen reduction process31. In the case of Fe-Cu@NC, the average charge transfer for Fe and Cu are 1.12 and 0.91, respectively. The findings indicate that the presence of Fe sites could promote the adsorption or activation of O2 molecules, thereby indirectly affecting the behavior of intermediates on Cu sites. In essence, taking into account interactions with both Fe and Cu sites offers a more comprehensive insight into the catalytic mechanism50.
Free energy
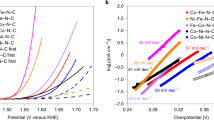
In this work, we considered the four-electron pathway for ORR in alkaline conditions to determine which catalyst has a lower overpotential (ηORR). First, we performed the calculations for two gas and solvent phases to evaluate the reaction path on different electrocatalysts. For the ideal catalyst at U = 0.4 V vs. SHE, the four steps have ΔGi = 0 and η = 0. The rate determination step (RDS) with a positive ΔG value is thermodynamically difficult to occur and leads to a high overpotential50,51. According to Fig. 2 and Table 2, *OOH formation is the most difficult step to occur on Fe@NC and Fe-Cu@NC in two gas and solvent phase systems. In contrast, the formation of *O from *OOH and the formation of *OOH are the rate determination steps for Cu@NC in the gas and solvent phases, respectively. In addition, Fig. 3 shows the free energy change of all catalysts at different potentials and pH = 14, according to Eqs. (11)–(14) in the gas phase. According to Eq. (16), the calculated UL values for Fe@NC, Cu@NC, and Fe-Cu@NC are −0.21, −0.31, and −0.26 V, vs. SHE, respectively. Also, the calculated overpotential values from Eq. (17) for Fe@NC, Cu@NC, and Fe-Cu@NC in the gas phase are 0.69 V, 0.71 V, and 0.66 V, respectively. Figure S3 shows the free energy change of all catalysts at different potentials and pH = 14, according to Eqs. (11)–(14) in the solvent phase. The calculated UL values Fe@NC, Cu@NC, and Fe-Cu@NC are −1.35, −1.40, and −1.17 V, vs. SHE, respectively. The calculated overpotential values of Fe@NC, Cu@NC, and Fe-Cu@NC in the solvent phase are 1.75 V, 1.80 V, and 1.57 V, respectively. Therefore, the results in the solvent phase lead to a higher overpotential and thermodynamic limiting potential. It is also clear that the ORR kinetic offered by Fe-Cu@NC is suitable and has been identified as the best candidate among these electrocatalysts, which also showed the major impression resulting from the carbon substrate and N doping.

Gibbs free energy diagrams of ORR in gas and solvent phases for: (a) Fe@NC, (b) Cu@NC, (c) Fe-Cu@NC models considered at U = 0 V and pH = 14.

Gibbs free energy diagrams of ORR in the gas phase for: (a) Fe@NC, (b) Cu@NC, (c) Fe-Cu@NC models considered at different electrode potentials and pH = 14, at the equilibrium potential (U0 = 0.4 V) and at UL (V vs. SHE), which is system dependent.
Conclusion
To increase the activity and performance of M–N/C catalysts, different strategies such as single-atom catalysts (SACs) and dual-atom catalysts can be used. Density functional theory calculations were employed to investigate the ORR activity of low-cost single Fe and Cu-doped and co-doped graphene-based structures in alkaline media as electrocatalysts of Oxygen-depolarized cathodes (ODCs) in advanced chlor-alkali electrolysis. In these catalytic models, the metal atoms are anchored at the hollow sites of N-doped graphene and each of them is coordinated to four nitrogen atoms. The four-electron ORR mechanism is investigated in the gas and solvent phases. The Gibbs free-energy profiles for different catalysts show that the results in the solvent phase lead to a higher overpotential and thermodynamic limiting potential. Among studied catalysts, Fe-Cu@NC is predicted to be the best candidate for cathode with the low thermodynamic limiting potential and theoretical overpotential of −0.26 and 0.66 V vs. SHE, respectively, at pH = 14 in the gas phase. The results indicated that the bimetallic synergy between Fe and Cu in dual atom-doped graphene facilitated the ORR in alkaline media. In fact, DFT calculations in this work helped us to investigate the reaction mechanism for single-atom and dual-atom catalysts, estimate the adsorption energies of chemical species, determine the activation energy barriers of the reactions, and provide information about the electronic structure. This study can be a guide for the design and discovery of new catalysts for oxygen reduction reaction in alkaline media in the future.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Ehelebe, K. et al. Fuel cell catalyst layer evaluation using a gas diffusion electrode half-cell: Oxygen reduction reaction on Fe-N-C in alkaline media. J. Electrochem. Commun. 116, 106761 (2020).
Wan, W. et al. Waste biomass derived active carbon as cost-effective and environment-friendly cathode material for lithium-oxygen batteries. J. Electrochem. Soc. 168, 050542 (2021).
Moussallem, J., Jörissen, U., Kunz, S. & Pinnow, Th. Turek, Chlor-alkali electrolysis with oxygen depolarized cathodes: History, present status and future prospects. J. Appl. Electrochem. 38, 1177–1194 (2008).
Baitalow, K., Köller, N., Bacmeister, P., Keller, R. & Wessling, M. On the operation of switchable oxygen depolarized cathodes. J. Chem. Eng. 469, 143759 (2023).
Sun, Ch. et al. Cobalt sulfides constructed heterogeneous interfaces decorated on N, S-codoped carbon nanosheets as a highly efficient bifunctional oxygen electrocatalyst. J. Mater. Chem. A. 9, 13926–13935 (2021).
Thanh, T. D., Chuong, N. D., Hien, H. V., Kim, N. H. & Lee, J. H. CuAg@Ag core-shell nanostructure encapsulated by N-doped graphene as a high-performance catalyst for oxygen reduction reaction. ACS Appl. Mater. Interfaces. 10, 4672–4681 (2018).
Xiao, H., Cheng, T. & Goddard, W. A. III. Atomistic mechanisms underlying selectivities in C1 and C2 products from electrochemical reduction of CO on Cu(111). J. Am. Chem. Soc. 139, 130–136 (2017).
Wang, Y., Chen, K. S. & Mishler, J. A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research. J. Appl. Energy. 88, 981–1007 (2011).
Huang, H. et al. Achieving remarkable activity and durability toward oxygen reduction reaction based on ultrathin Rh-doped Pt nanowires. J. Am. Chem. Soc. 139, 8152–8159 (2017).
Jain, D. et al. Experimental and DFT investigation into chloride poisoning effects on nitrogen-coordinated iron-carbon (FeNC) catalysts for oxygen reduction reaction. J. Phys. Chem. C. 124, 10324–10335 (2020).
Shao, M., Chang, Q. & Dodelet, J. P. Recent advances in electrocatalysts for oxygen reduction reaction. J. Chem. Rev. 106, 3983–4021 (2016).
Gao, Y. et al. Energy density-enhancement mechanism and design principles for heteroatom-doped carbon supercapacitors. J. Nano Energy. 72, 104666 (2020).
Tang, W. et al. Recent advances of bifunctional catalysts for zinc air batteries with stability considerations: From selecting materials to reconstruction. Nanosc. Adv. 5, 4368–4401 (2023).
Tian, X., Feng Lu, X., Yu Xia, B. & Wen Lou, X. Advanced electrocatalysts for the oxygen reduction reaction in energy conversion technologies. J. Joule. 4, 45–68 (2020).
S. Kolluru, G. Mahnot Jain, D. Gollapudi, L. Eswaraditya Reddy, G. Ramesh (2023) Recent progress in the development of Platinum-based electrocatalysts for the oxidation of ethanol in fuel cells. J. Mater. Today: Proc.
Sun, X. et al. Self-supporting metal–organic framework-based hydrogen and oxygen electrocatalysts. J. Mater. Chem. A. 11, 13089–13106 (2023).
Zhang, D. et al. Metal-free carbon-based catalysts design for oxygen reduction reaction towards hydrogen peroxide: From 3D to 0D. J. Mater. Today. 63, 339–359 (2023).
Song, M. et al. Single-atom catalysts for H2O2 electrosynthesis via two-electron oxygen reduction reaction. J. Adv. Funct. Mater. 33, 2212087 (2023).
Fu, Sh., Zhu, Ch., Song, J., Du, D. & Lin, Y. Metal-organic frameworks based porous carbons for oxygen reduction reaction electrocatalysts for fuel cell applications. J. Nanocarbon Electrochem. 8, 1700363 (2019).
Deshpande, S., Kitchin, J. R. & Viswanathan, V. Quantifying uncertainty in activity volcano relationships for oxygen reduction reaction. J. ACS Catal. 6, 5251–5259 (2016).
Cheng, D., Qiua, X. & Yu, H. Enhancing oxygen reduction reaction activity of Pt-shelled catalysts via subsurface alloying. J. Phys. Chem. Chem. Phys. 16, 20377–20381 (2014).
Duan, Z. & Wang, G. Comparison of reaction energetics for oxygen reduction reactions on Pt (100), Pt (111), Pt/Ni (100), and Pt/Ni (111) surfaces: A first-principles study. J. Phys. Chem. C. 117, 6284–6292 (2013).
Anderson, A. B., Jinnouchi, R. & Uddin, J. Effective reversible potentials and onset potentials for O2 electroreduction on transition metal electrodes: Theoretical analysis. J. Phys. Chem. C. 117, 41–48 (2013).
Iyemperumal, S. K. & Deskins, N. A. Evaluating solvent effects at the aqueous/Pt (111) interface. J. ChemPhysChem. 18, 2171–2190 (2017).
J.A. Gauthier, C.F. Dickens, L.D. Chen, A.D. Doyle, J.K. Norskov. Solvation effects for oxygen evolution reaction catalysis on IrO2(110). J. Phys. Chem. C., (2017).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B. 108, 17886–17892 (2004).
Zhang, Y., Zi, Su., Wei, H., Wang, Zh. & Gong, X. Strategies to improve the oxygen reduction reaction activity on Pt–Bi bimetallic catalysts: A density functional theory study. J. Phys. Chem. Lett. 14, 1990–1998 (2023).
Gao, Q. A DFT study of the ORR on M-N3 (M = Mn, Fe Co, Ni, or Cu) co-doped graphene with moiety-patched defects. J. Ionics. 26, 2453–2465 (2020).
Yu, L., Pan, X., Cao, X., Hu, P. & Bao, X. Oxygen reduction reaction mechanism on nitrogen-doped graphene: A density functional theory study. J. Catal. 282, 183–190 (2011).
Ignaczak, R. et al. Schmickler, oxygen reduction in alkaline media—A discussion. J. Electrocatalysis. 8, 554–564 (2017).
Jangjooye Shaldehi, T. et al. Computationally screening non-precious single atom catalysts for oxygen reduction in alkaline media. J. Catal. Today 431, 114560 (2024).
Liang, Q., Brocks, G. & Bieberle-Hütter, A. Oxygen evolution reaction (OER) mechanism under alkaline and acidic conditions. J. Phys. Energy. 3, 026001 (2021).
Man, H. et al. Rossmeisl, Universality in oxygen evolution electrocatalysis on oxide surfaces. J. Chemcatchem. 3, 1159–1165 (2011).
López, M., Exner, K., Viñes, F. & Illas, F. Theoretical study of the mechanism of the hydrogen evolution reaction on the V2C MXene: Thermodynamic and kinetic aspects. J. Adv. Theory Simul. 6, 2200217 (2023).
Exner, K. S., Anton, J., Jacob, T. & Over, H. Controlling selectivity in the chlorine evolution reaction over RuO2-based catalysts. Electrochim Acta. 120, 460 (2014).
López, M., Exner, K. S., Viñes, F. & Illas, F. Computational Pourbaix diagrams for MXenes: A key ingredient toward proper theoretical electrocatalytic studies. Adv. Theory Simul. 6, 2200217 (2022).
Kulkarni, S. S., Patel, A. & Nørskov, J. K. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 118, 2302–2312 (2018).
Delley, From molecules to solids with the DMol3 approach. J. Chem. Phys. 113, 7756–7764 (2000).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. J. Phys. Rev. Lett. 77, 3865–3868 (1996).
Delley, B. Hardness conserving semilocal pseudopotentials. J. Phys. Rev. B Condens. 66, 155125 (2002).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Delley, B. The conductor-like screening model for polymers and surfaces. J. Mol. Simul. 32, 117–123 (2006).
Posada-Pérez, S. et al. CO2 interaction with violarite (FeNi2S4) surfaces: A dispersion-corrected DFT study. J. Phys. Chem. Chem. Phys. 20, 20439–20446 (2018).
Kattel, S. & Wang, G. A density functional theory study of oxygen reduction reaction on Me–N4 (Me = Fe Co, or Ni) clusters between graphitic pores. J. Mater. Chem. A. 1, 10790 (2013).
Yan, M. et al. Single-iron supported on defective graphene as efficient catalysts for oxygen reduction reaction. J. Phys. Chem. C. 24, 13283–13290 (2020).
Vinogradov, K. Y. et al. Density functional theory study of the oxygen reduction reaction mechanism on graphene doped with nitrogen and a transition metal. J. ACS Omega. 7, 7066–7073 (2022).
Li, Zh. et al. Engineering d-band center of FeN4 moieties for efficient oxygen reduction reaction electrocatalysts. J. Energy Storage Mater. 59, 102764 (2023).
Tang, W., Sanville, E. & Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Compute. Mater. 21, 084204 (2009).
Sanville, E., Kenny, S. D., Smith, R. & Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comp. Chem. 28, 899–908 (2007).
Xu, J., Elangovan, A., Li, J. & Liu, B. Graphene-based dual-metal sites for oxygen reduction reaction: A theoretical study. J. Phys. Chem. C. 125(4), 2334–2344 (2021).
Koper, M. T. M. Analysis of electrocatalytic reaction schemes: Distinction between rate-determining and potential-determining steps. J. Solid State Electrochem. 17, 339–344 (2013).
Acknowledgements
The authors are thankful to Prof. Francesc Illas for fruitful discussions regarding the DFT calculations of oxygen reduction reaction in alkaline media. This research is financially supported by the Iran National Science Foundation (INSF-4005715).
Author information
Authors and Affiliations
Contributions
Tahereh Jangjooye Shaldehi: Conceptualization Data curation Formal analysis Investigation Validation Visualization Writing - original draft Soosan Rowshanzamir: Methodology Project administration Resources Software Supervision Validation Visualization Writing - review & editing
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shaldehi, T.J., Rowshanzamir, S. Theoretical investigation of electrocatalytic activity of Pt-free dual atom-doped graphene for O2 reduction in an alkaline solution. Sci Rep 14, 14201 (2024). https://doi.org/10.1038/s41598-024-61223-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-61223-y
Keywords
This article is cited by
-
Versatile single atom-perovskite materials: synthesis, characterization, applications, and perspectives
NPG Asia Materials (2025)
