
Abstract
Cancer therapy-related cardiac dysfunction (CTRCD), which commonly includes left ventricular dysfunction and heart failure, is the main adverse effect of anticancer therapy. In recent years several candidate genes studies and genome-wide association studies have identified common genetic variants associated with CTRCD, but evidence remains limited and few genetic variants are robust. A genome-wide meta-analysis of CTRCD was performed with 852 oncology patients receiving cancer therapy. DNA samples were genotyped and imputed to perform a GWAS meta-analysis for case–control (N = 852 (380 cases and 472 controls) and extreme phenotypes (N = 618 (78 cases and 472 controls) looking for genetic variants that predispose to CTRCD. The results were validated in a replicate cohort of 1,191 oncology patients (245 cases and 946 controls). Functional mapping of the replicated loci was then performed. The meta-analysis showed 9 and 17 loci suggestively associated (P-value < 1 × 10–5) with CTRCD in case–control and extreme phenotypes analyses, respectively. The 3q28 locus (rs rs7652759, P = 5.64 × 10–6) in the case–control analysis was the strongest signal, with up to 64 SNPs above the suggestive significance threshold. The rs7652759, an intergenic variant between TPRG1 and TP63 genes, was the only variant validated in the replication cohort (P-value = 0.01). Functional mapping of this significant locus revealed up to 5 new genes potentially involved in the CTRCD. We identified the intergenic region near TP63 as a novel CTRCD susceptibility locus. In the future, the genotyping of these markers could be considered in new CTRCD risk scores to improve preventive strategies in cardio-oncology.
Similar content being viewed by others
Introduction
The survival of cancer patients has increased considerably in the last decades, exceeding 80% of cases, with variations depending on the type of cancer and the country. One of the current challenges is adverse events in cancer treatment, that can lead to the discontinuation of effective therapies1 and affect the quality of life and longevity of cancer survivors, especially, cancer therapy-related cardiovascular toxicity. The most relevant cardiovascular side effects associated with these therapies are left ventricular dysfunction (LVD) and heart failure (HF)2,3 which we will refer to as cancer therapy-related cardiac dysfunction (CTRCD).
The correct management of CTRCD is essential to identify high-risk patients before the start of cancer therapy, to follow a proper prevention and surveillance plan for early identification. Existing evidence supports the hypothesis that genetic variation contributes to CTRCD susceptibility, so the identification of genetic variants could improve risk assessment. In recent years, several candidate genes and genome-wide association studies have identified common genetic variants associated with CTRCD, but the evidence is still limited, and only a few genetic variants are robust4,5. Rare variants in genes that cause familial cardiomyopathies have also been identified6. Future studies are necessary for validation and assessment of their value in a diagnostic and prognostic setting.
In this study, we carry out one of the largest CTRCD meta-analysis to date to identify genetic variants associated with this phenotype. Genotyping of these common variants, along with rare variants and clinical variables, would allow the development of a risk predictor model to provide an estimate of an individual's risk of CTRCD before starting cancer therapy.
Results
Discovery GWAS for CTRCD
We use a genome-wide association approach to search for susceptibility genes for CTRCD in patients undergoing cancer treatment. No loci exceeded the conventional genome-wide significance (P-value ≤ 5.0 × 10−8) in any of the analyses, but 8 loci for case–control and 22 loci for extreme-phenotypes analysis exceeded suggestive significance (P-value ≤ 1.0 × 10−5). In case–control analysis, the two strongest associations with CTRCD were at the 19q13.32 locus, near EML2 gene (rs12463090, P = 5.26 × 10–7) and at the 13q12.3 locus, near MTUS2 gene (rs35858036, P = 5.85 × 10–7). In extreme phenotypes despite having 22 suggestive loci, the signals were less robust, probably due to the small sample size. The higher signal in this analysis was in the LOC339166 gene, at the locus 17p13.2 (rs7208927, P = 2.06 × 10–6).
The 2022 ESC guidelines on cardio-oncology2 describes several genetic variants that drives CTRCD (CELF4 rs1786814; RARG rs2229774; SLC28A3 rs7853758; UGT1A6 rs17863783; and one intergenic variant, rs28714259). We tested these variants in our cohort, and only the intergenic variant rs28714259 showed hints of suggestive association (P-value = 1 × 10–3, in case–control analysis).
Meta-analysis for CTRCD
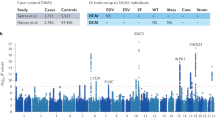
To enhance the power of genetic discovery, we meta-analyzed our GWAS summary statistics with two additional cohorts. In the case–control GWAS meta-analysis, 9 loci exceeded the suggestive significance. The rs4803842 achieved the highest probability (P = 9.67 × 10–8). This variant is at the 19q13.32 locus, which is consistent with the results of the discovery GWAS. However, the most robust signal from the meta-analysis was found in an intergenic region on chromosome 3 (rs7652759, P = 5.64 × 10–6), with up to 64 SNPs exceeding the threshold of suggestive significance. Regarding the extreme-phenotypes analysis, the two main signals were found on chromosomes 6 (rs77245823, P = 1.12 × 10–6) and 11 (rs78367094, P = 1.18 × 10–6) (Fig. 1).

Miami plot showing the P-values for GWAS-meta-analysis for case–control analysis (top) and extreme phenotypes (bottom). Only variants shared between the three cohorts are represented. The orange horizontal line indicates suggestive significance for common variants at − log10(1 × 10−5).Replication of the significant SNPs of the meta-analysis.
To strengthen the results, we tested the SNPs that exceeded suggestive significance in the meta-analysis, after LD cumpling, in the N9831 cohort (Table 1). The rs7652759, an intergenic variant between TPRG1 and TP63 genes (Fig. 2A), was the only variant validated in the replication cohort (P-value = 1.08 × 10–2). This is probably due to limited sample size or differences in cohort characteristics.

Regional plot and chromatin interactions of the locus 3q28 of the CTRCD meta-analysis. (A) Zoom in on the regional plot of the TP63 locus, which includes prioritized genes TP63, LEPREL1, KNG1, MASP1 and CLDN1. Genes prioritized by FUMA are highlighted in red. (B) Circo plot showing genes on chromosome 3 that were implicated through the genomic risk loci (blue area) by chromatin interaction mapping (orange font). The outer layer shows a Manhattan plot containing the –log10-transformed P value of each SNP in the meta-analysis. Empty regions in the Manhattan plot layer indicate regions where no SNPs with P < 0.05 were situated.
Gene prioritization at the 3q28 locus highlighted novel genes likely involved in CTRCD
After confirming the association of rs37652759 with the CTRCD, we performed a functional mapping to prioritise a set of candidate genes at this locus. Positional and eQTL mapping in the 3q28 locus revealed no significant association with any gene. However, chromatin interaction mapping using Hi-C data from cardiac tissue identified 5 genes (KNG1, MASP1, TP63, LEPREL1 and CLDN1), most of which were physically located outside the risk locus (Fig. 2B). These results include new candidate genes involved in CTRCD that have not been previously reported.
Discussion
Over the last few years, several efforts have been made to search for genetic variants that allow us to identify individuals with susceptibility to develop CTRCD. Candidate gene approaches and GWAS studies have pointed out some genetic variants of potential interest, but in most cases based on small and/or heterogeneous cohorts. This is the first association study with a significant number of participants, with clear and standardized criteria for the classification of cardiotoxicity, focused on CTRCD. We here provide evidence of the potential involvement of specific genetic alterations, such as the intergenic variant rs7652759, and 5 candidate genes in CTRCD susceptibility.
One of the genes identified by functional mapping in our study (KNG1) was previously associated with doxorubicin cardiotoxicity, which reinforces its validity. In particualr, Cheng et al.7 showed that KNG1 overexpression aggravates mitochondrial dysfunction in doxorubicin heart injury. Following on from mitochondrial dysfunction, it has recently been pointed out that p53 prevents doxorubicin cardiotoxicity due to its protective effect on the mitochondrial genome, independently of its prototypical tumor suppressor activity8,9,10. In our study, we found an association of CTRCD with the TP63 gene, which is a member of the p53 family of transcription factors, and could therefore be expected to have a similar function.
On the other hand, of the remaining genes, LEPREL1 (also called P3H2) has been associated with collagen IV11, the major and crucial component of basement membranes; and CLDN112,13 and MASP114 with fibrosis, a key driver of end-stage organ failure and cancer.
In summary, functional mapping revealed up to 5 candidate genes involved in multiple steps of CTRCD pathophysiology (4 of them previously undescribed). Although our results are in agreement with previous literature, further genetic and functional studies are needed to elucidate the actual involvement of these genes in the development of CTRCD.
Limitations
The main limitation of this study is the sample size, which made it difficult to achieve significant statistical power. However, our approach used the whole genome-wide set of summary statistics from 3 different countries, which strengthened the results and allowed us to detect potential susceptibility loci that did not reach suggestive significance in those studies by themselves. Despite this, our study is one of the largest meta-analyses of susceptibility to CTRCD to date. Another possible limitation is the existence of small variations in the CTRCD criteria in the added series.
Conclusion
In summary, this study identified the rs7652759 and 5 novel putative CTRCD susceptibility genes. These findings provide a guide for further experimental validation of functional variants and disease-related genes, and help us to understand better the pathophysiological mechanisms that cause cardiotoxicity. In the future, a personalized genetic approach may help to better define individual susceptibility to CTRCD, thus improving the quality of life and survival of cancer patients.
Methods
Discovery phase
The cohort
The discovery cohort included 620 adult patients from the CARDIOTOX registry who were followed for a median of 24 months. According to the worst myocardial injury/dysfunction observed during the CARDIOTOX registry follow-up, patients were classified as follows15: (1) Controls (N = 325), if they show normal left ventricular function and biomarkers (high-sensitivity troponin T and N-terminal natriuretic pro-peptide); (2) Mild cases (N = 224), if they presented with LVEF > 50% with elevated biomarkers or at least one additional abnormal echo parameter; (3) Moderate cases (N = 22), asymptomatic patients with LVEF ≥ 40% and < 50%; (4) Severe cases (N = 49), patients with asymptomatic LVEF < 40% or clinical HF.
Information regarding age, gender, primary tumor type, use of radiotherapy, cardiovascular history, body mass index, smoking, arterial hypertension, sedentary lifestyle, diabetes, hypercholesterolemia, and not a healthy diet, was obtained from the CARDIOTOX prospective database. Detailed information about the clinical data is available in Table 2. The ethics committee of the La Paz University Hospital approved the study (PI-2389) and all participants gave their written informed consent. Furthermore, research was performed in accordance with relevant guidelines/regulations.
Genotyping and imputation
Individuals of the discovery cohort were genotyped at the Spanish National Genotyping Center (CeGen-FPGMX; Santiago de Compostela, Spain), using the Axiom™ Spain Biobank array and following strictly the manufacturer's instructions (Axiom™ 2.0 Assay 96-Array Format Manual Workflow; ThermoFisher Scientific).
A quality control pipeline was run in PLINK. Variants with genotyping rate < 98% and MAF < 1% were removed. Individuals with > 2% missing genotypes were excluded from further analyses. LD-pruning was performed before Identity-By-Descent calculation and PCA. IBD was examined to identify and exclude potential relatives up to a third degree. Likewise, subsequent rounds of outlier identification according to genomic principal components (PCs) were run to evaluate potential stratification and select a subset of the homogeneous sample.
Genotypes were phased and imputed using Eagle v2.4 and minimac4, respectively, in the Michigan Imputation Server. The 1000 Genomes Phase 3 v5 was used as a reference panel (array built by GRCh37/hg19). Post-imputation quality control involved the exclusion of multiallelic SNPs and indels, as well as SNPs with a low imputation score, low MAF (< 1%), and high missingness (> 2%). After imputation procedures, 573 individuals (280 cases and 293 controls) and 8,567,906 SNPs on chromosomes 1–22 remained for the analysis. Figure 3 shows a scheme of the main steps followed for the study.

Analysis pipeline. Shows the main steps followed during the research process.
Genome-wide association analysis
Association testing was performed by fitting logistic regression models in PLINK 2.0 under an additive model with age, sex, and principal component 1 (PC1) as covariables. Variants with P-value < 5 × 10–8 and P-value < 1 × 10–5 were considered significant and suggestive, respectively. We performed a GWAS for case–control and extreme-phenotypes analysis, considering patients with CTRCD and severe CTRCD, respectively, as cases. In total, 280 cases and 293 controls for case–control analysis and 46 cases and 293 controls for extreme-phenotypes analysis. The Manhattan plot was plotted using the R package qqman (https://github.com/stephenturner/qqman)16.
Meta-analysis of CTRCD
Description of the cohorts
To maximize the statistical power and detect associated genetic variants of small effect, we added 2 independent cohorts to the discovery cohort and performed meta-analyses for case–control and extreme phenotypes in METAL17.
Specifically, a total of 227 female breast cancer patients treated with epirubicin from the University Hospital Leuven (Leuven, Belgium), and 52 female breast cancer patients treated with doxorubicin from Hospital Clínico San Carlos (Madrid, Spain) were included. Patients in both cohorts were reclassified according to the discovery cohort criteria for cardiotoxicity. Patients had normal cardiac function before the chemotherapy treatment. The logistic regression was adjusted for anthracyclines cumulative dose. Detailed information on the cohorts can be found in the previously published article18.
Meta-analysis procedure
In total, 852 individuals (380 cases and 472 controls) for case–control and 618 individuals (78 cases and 472 controls) for extreme phenotypes analysis were included in the meta-analysis. Meta-analysis was performed using a whole genome-wide set of summary statistics. Only variants shared between cohorts were considered. Meta-analysis was performed by weighting the effect size estimates using the inverse of the standard errors. The P-value and direction of effect are weighted according to sample size of each cohort. Heterogeneity between studies was evaluated with the Cochran’s-Q test. Variants showing heterogeneity of effect between the datasets were excluded. A quantile–quantile (QQ) plot was generated using the observed and expected P-values. The genomic inflation factor (lambda), calculated based on the 50th percentile, was 0.955 for case–control and 1.012 for extreme phenotypes analysis, indicating no significant population stratification.
Replication of significant SNPs
LD clumping was performed on SNPs that passed the threshold of suggestive significance (P < 1 × 10–5) in the meta-analysis, and subsequently were tested in the N9831 cohort. A SNP was considered to replicate if the probability exceeds the threshold P < N SNPs/0.05. The N9831 replicate cohort is described below. This cohort included 1191 HER2 + breast cancer patients from The North Central Cancer Treatment Group (NCCTG) N9831 clinical trial, which were collected in the United States. These patients were treated with doxorubicin, cyclophosphamide and paclitaxel, or doxorubicin, cyclophosphamide, paclitaxel, and trastuzumab. The N9831 cohort used the definition of cases as per the Gomez-Peña article on trastuzumab cardiotoxicity19: A decrease of at least 10% from baseline with a resulting LVEF of less than 50% at follow-up, a decrease of 15% concerning LVEF baseline, or any decrease resulting in LVEF less than 45% at least once during the treatment or clinical manifestation of congestive heart failure. For extreme phenotype, the definition was: patients with cardiac events, defined as symptomatic HF, definite cardiac death (CD; as a result of myocardial infarction, HF, or arrhythmia), or probable CD (patient death without documented etiology). Three cardiologists independently investigated all cardiac events, and an event was confirmed as a cardiac event if agreement was reached between at least two cardiologists20. Briefly, individuals with a non-white or Hispanic origin and individual DNA samples and SNPs that failed quality control were excluded. The logistic regression was adjusted for age, baseline LVEF, anti-hypertensive medications, and the first two principal components. Detailed information about the cohort can be found in the previously published article21.
Characterization of genomic risk loci and functional annotation
Functional annotation was performed with FUMA22, an online platform for the functional mapping of genetic variants. First, SNPs with a suggestive significant P value (< 1 × 10−5) and independent from each other at r2 < 0.6 and r2 < 0.1 are identified as independent significant SNPs and lead SNPs, respectively. All known SNPs that have r2 ≥ 0.6 with one of the independent significant SNPs are included for further annotation (candidate SNPs). Subsequently, genomic risk loci were defined by merging independent significant SNPs that are < 250 kb located to each other. Each genomic risk loci can thus contain multiple independent significant SNPs and lead SNPs. The rationale behind this approach is that the most significant SNP at the locus is not necessarily the causal SNP, but rather could be in LD with the causal SNP22. 1000 Genomes Europe was used as a reference panel.
Functional consequences for these SNPs on gene functions (based on Ensembl genes)23, deleteriousness score (Combined annotation-dependent depletion-CADD score, with scores > 12.37 seen as likely deleterious)24, potential regulatory information such as enhancer/promoter regions (RegulomeDB (RDB) scores, for which a higher probability of having a regulatory function is indicated by a lower score)25, effects on gene expression using eQTLs of GTEx26 cardiac tissues and 3D structure of chromatin interactions with High-throughput Chromosome Conformation Capture (Hi-C) data were annotated with ANNOVAR to each SNP23.
Gene mapping
Functional annotated SNPs are subsequently mapped to genes based on functional consequences on genes by three strategies. First, positional mapping is performed using the physical distances (within 20-kb windows) from known protein-coding genes in the human reference assembly (GRCh37)22. A CADD score threshold of 12.37 and RDB score < 3 were used for filtering SNPs, to prioritize SNPs that are likely to have a deleterious effect or to affect regulatory elements, respectively. Second, for eQTL mapping we used information on cardiac tissues from GTEx v7 to map SNPs to genes up to 1 Mb away (cis-eQTLs) based on a significant eQTL association (false discovery rate (FDR) ≤ 0.05), which indicates that the expression of that gene is associated with allelic variation at the SNP. Third, chromatin interaction mapping mapped SNPs to genes based on a significant chromatin interaction between a genomic region in a risk locus and promoter (250 bp upstream and 500 bp downstream of a transcription start site (TSS) and enhancer regions of genes22. FUMA uses Hi-C data from cardiac tissues. This type of mapping does not have a distance boundary and may therefore involve long-range interactions. Similar to eQTL mapping, we used an FDR of 1 × 10−5 to define significant interactions.
The three mapping strategies result in a set of prioritized genes. Both eQTL and chromatin interaction mapping may lead to prioritized genes that are not necessarily located inside a genomic risk locus, although they are linked to SNPs within a genomic risk locus22. The combination of positional mapping of deleterious SNPs, eQTL mapping, and chromatin interaction mapping across cardiac tissues may reveal multiple lines of evidence pointing towards the same genes and enables to prioritize genes that are highly likely involved in CTRCD22.
Ethical approval
Local institutional review boards (PI-2389) approved the study ethically, and all participants provided written informed consent.
Data availability
Data is provided within the manuscript or supplementary information files.
Abbreviations
- AE:
-
Effect allele
- CADD:
-
Combined annotation-dependent depletion
- CTRCD:
-
Cancer therapy-related cardiac dysfunction
- eQTL:
-
Expression of quantitative trait locus
- FDR:
-
False discovery rate
- GWAS:
-
Genome-wide association study
- HF:
-
Heart failure
- HWE:
-
Hardy-Weinberg equilibrium
- IBS:
-
Identity by state
- IndSigSNP:
-
Independent significant SNP
- LD:
-
Linkage disequilibrium
- LVEF:
-
Left ventricular ejection fraction
- MAF:
-
Minor allele frequency
- ncRNA:
-
Non-Coding RNA
- QC:
-
Quality control
- QQ plot:
-
Quantile-quantile plot
- RDB:
-
Regulome database
References
Sardesai, S. et al. Clinical impact of interruption in adjuvant Trastuzumab therapy in patients with operable HER-2 positive breast cancer. Cardiooncology. 6, 26 (2020).
Lyon, A. R. et al. 2022 ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 43, 4229–4361 (2022).
Plana, J. C. et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 27, 911–939 (2014).
Al-Otaibi, T. K., Weitzman, B., Tahir, U. A. & Asnani, A. Genetics of anthracycline-associated cardiotoxicity. Front Cardiovasc. Med. 9, 867873 (2022).
Berkman, A. M., Hildebrandt, M. A. T. & Landstrom, A. P. The genetic underpinnings of anthracycline-induced cardiomyopathy predisposition. Clin. Genet. 100, 132–143 (2021).
Garcia-Pavia, P. et al. Genetic variants associated with cancer therapy-induced cardiomyopathy. Circulation. 140, 31–41 (2019).
Cheng, X. et al. Overexpression of Kininogen-1 aggravates oxidative stress and mitochondrial dysfunction in DOX-induced cardiotoxicity. Biochem. Biophys. Res. Commun. 550, 142–150 (2021).
Li, J. et al. p53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc. Natl. Acad. Sci. U S A. 116, 19626–19634 (2019).
Nishi, M., Wang, P.-Y. & Hwang, P. M. Protective role of p53 in doxorubicin-induced cardiomyopathy as a mitochondrial disease. Mol. Cell Oncol. 7, 1724598 (2020).
McSweeney, K. M., Bozza, W. P., Alterovitz, W.-L. & Zhang, B. Transcriptomic profiling reveals p53 as a key regulator of doxorubicin-induced cardiotoxicity. Cell Death Discov. 5, 102 (2019).
Pokidysheva, E. et al. Biological role of prolyl 3-hydroxylation in type IV collagen. Proc. Natl. Acad. Sci. 111, 161–166 (2014).
Roehlen, N. et al. A monoclonal antibody targeting nonjunctional claudin-1 inhibits fibrosis in patient-derived models by modulating cell plasticity. Sci. Transl. Med. 14, eabj4221 (2022).
Zou, J. et al. Idiopathic pulmonary fibrosis is associated with tight junction protein alterations. Biochim. Biophys. Acta Biomembr. 1862, 183205 (2020).
Hess, K. et al. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS One. 7, e35690 (2012).
López-Sendón, J. et al. Classification, prevalence, and outcomes of anticancer therapy-induced cardiotoxicity: the CARDIOTOX registry. Eur. Heart J. 41, 1720–1729 (2020).
Turner, D. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. JOSS. 3, 731 (2018).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 26, 2190–2191 (2010).
Velasco-Ruiz, A. et al. POLRMT as a novel susceptibility gene for cardiotoxicity in epirubicin treatment of breast cancer patients. Pharmaceutics. 13, 1942 (2021).
Gómez Peña, C. et al. Influence of the HER2 Ile655Val polymorphism on trastuzumab-induced cardiotoxicity in HER2-positive breast cancer patients: a meta-analysis. Pharmacogenet. Genomics. 25, 388–393 (2015).
Advani, P. P., Ballman, K. V., Dockter, T. J., Colon-Otero, G. & Perez, E. A. Long-term cardiac safety analysis of NCCTG N9831 (alliance) adjuvant trastuzumab trial. J. Clin. Oncol. 34, 581–587 (2016).
Serie, D. J. et al. Genome-wide association study of cardiotoxicity in the NCCTG N9831 (Alliance) adjuvant trastuzumab trial. Pharmacogenet. Genomics. 27, 378–385 (2017).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–5 (2013).
Acknowledgements
We express our gratitude to the patients and their families for their participation in the study. We thank Fundación Pública Galega de Medicina Xenómica (FPGMX) for the provision of the genotyping (Cegen) and bioinformatic facilities and team.
Funding
This work was supported by Instituto de Salud Carlos III (ISCIII) –– “Next Generation EU” Funds – Mecanismo de Recuperación y Resiliencia (MRR) (PMP22/00098, PI19/01283, PI18/01242 and PI13/00559). Ph.D. Student Martínez-Campelo L had a Predoctoral Contract for Training in Health Research from Carlos III Health Institute.
Author information
Authors and Affiliations
Consortia
Contributions
M.B., A.C. and J.L.L.S. designed the study. L.M. analyzed and interpreted the data and wrote the manuscript. A.B., R.C., and S.D., participated in the analysis and interpretation of the data. N.N., J.S.R., A.V., and A.G., performed the analysis of their respective cohorts. J.L.L.S., J.R.G.J., T.L.F., A.B., A.M.M., P.M., C.V., T.A.G., participated in the creation of the CARDIOTOX registry and C.V. and T.A.G. participated in the collection of additional cohorts. A.C. provided the facilities and equipment to perform the DNA extraction and genotyping of the samples. M.B. participated in the interpretation of the data and managed the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Martínez-Campelo, L., Blanco-Verea, A., López-Fernández, T. et al. Meta-analysis of genome-wide association studies for cancer therapy-related cardiovascular dysfunction and functional mapping highlight an intergenic region close to TP63. Sci Rep 14, 18413 (2024). https://doi.org/10.1038/s41598-024-69064-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-69064-5
