
Abstract
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) causes coronavirus disease 2019 (COVID-19), a pneumonia with extremely heterogeneous clinical presentation, ranging from asymptomatic to severely ill patients. Previous studies have reported links between the presence of host genetic variants and the outcome of the COVID-19 infection. In our study, we used whole exome sequencing in a cohort of 444 SARS-CoV-2 patients, admitted to hospital in the period October-2020–April-2022, to search for associations between rare pathogenic/potentially pathogenic variants and COVID-19 progression. We used gene prioritization-based analysis in genes that have been reported by host genetic studies. Although we did not identify correlation between the presence of rare pathogenic variants and COVID-19 outcome, in critically ill patients we detected known mutations in a number of genes associated with severe disease related to cardiovascular disease, primary ciliary dyskinesia, cystic fibrosis, DNA damage repair response, coagulation, primary immune disorder, hemoglobin subunit β, and others. Additionally, we report 93 novel pathogenic variants found in severely infected patients who required intubation or died. A network analysis showed main component, consisting of 13 highly interconnected genes related to epithelial cilium. In conclusion, we have detected rare pathogenic host variants that may have influenced the COVID-19 outcome in Bulgarian patients.
Similar content being viewed by others


Introduction
Since the first outbreak of the Coronavirus disease in the 2019 (COVID-19) pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), over 771 million cases of COVID-19 and more than 6.9 million deaths have been confirmed (https://covid19.who.int/, last accessed in November 2023). SARS-CoV-2 infection exhibits varying infectivity and mortality rates in different worldwide populations1. To date, fatality ratios (defined as the number of deaths divided by the number of confirmed cases) for the 20 countries most affected by COVID-19 worldwide range from 0.1% registered in South Korea to 4.9% reported in Peru (https://coronavirus.jhu.edu/data/mortality—accessed on 1st of April, 2024). At the time of writing (April 2024), there have been more than 1.3 million confirmed cases and 38,748 deaths in Bulgaria (case fatality rate of 2.9%) (https://covid19.who.int/region/euro/country/bg).
COVID-19 is a complex, highly infectious disease, with a highly variable presentation varying from the lack of symptoms, to severe disease with respiratory failure, overactive immune response and death. Generally, the first clinical presentations of the infection are similar to symptoms caused by other respiratory viruses such as influenza viruses, and include fever, cough, and fatigue. Less common signs comprise headache, sore throat, myalgia, arthralgia, diarrhea, vomiting, and changes in smell (anosmia, hyposmia) and taste (ageusia, dysgeusia)2. In severe cases, breathing difficulties develop, namely dyspnea, with acute respiratory distress syndrome being the most serious complication3. Approximately one-third of infected could be asymptomatic. Among symptomatic patients, 81% have mild to moderate symptoms, 14% develop severe symptoms (dyspnea, hypoxia, lung involvement on imaging); 5% experience respiratory failure, acute respiratory distress syndrome, shock, or multiorgan failure)4.
A common feature in critical illness is immune dysregulation which is due to hyperactivation of the NLRP3 inflammasome, leading to a sudden increase in proinflammatory cytokines and other inflammatory markers (cytokine storm). This hyperinflammatory syndrome causes thrombosis, coagulopathies, oxidative stress, multiorgan damage, and death5,6.
Major risk factors for severe disease complications (and death) include demographic characteristics (male sex and age above 65 years) and co-morbidities such as obesity7,8, hypertension, diabetes, chronic pulmonary, renal, respiratory or liver disease, immunodeficiencies, cancer, and cardiovascular disease, but these factors do not fully explain the wide range of clinical severity9,10. A possible explanation for these findings is that the host genetic variations affecting the structure or function of essential proteins play a critical role in the entry and spread of SARS-CoV-2 and determine the severity of COVID-19 course and outcome11.
Considering specificity of the host genome different strategies for genetic study of COVID-19 severity have been designed—studies which aimed the identification of: first, common single nucleotide polymorphisms (SNPs; usually with low effect size), and second, rare and ultra-rare variants (with higher impact) associated with different phenotypes. In the first case, strategies were directed from single-gene or candidate-pathway association studies, to genome-wide association studies (GWAS) and meta-analyses, attempting to build polygenic risk scores (The COVID-19 Host Genetics Initiative; The Severe Covid-19 GWAS Group; Genetics of Mortality in Critical Care initiative)12,13,14,15. In the second case, rare variants were identified through the implementation of next-generation sequencing (NGS) approaches, like sequencing of candidate-genes panels, as well as whole-exome and whole-genome sequencing experiments on large cohorts of patients and controls12,14,16,17. These studies were mostly performed by huge public consortia with different number of participating samples, and also by direct-to-consumer genetic companies (23andMe; https://www.23andme.com/; AncestryDNA; https://www.ancestry.com/dna/). The GWAS meta-analysis of the COVID-19 Host Genetics Initiative identified 23 loci with minor allele frequencies ranging from 0.003 to 0.66, of which seven loci influenced susceptibility to SARS-CoV-2 infection18, and 16 loci, including loci involved in inflammation or innate immunity (e.g., the OAS1/OAS2/OAS3 gene cluster, THBS3, SFTPD, MUC5B, ELF5, FBRSL1, SLC22A31, and NR1H2) were strongly associated with severe and critical disease (https://app.covid19hg.org/variants). The COVID-19 Host Genetics Initiative report agreed with the association of variants in ABO, SLC6A20, TYK2, DPP9, IFNAR2, and PPP1R15A in influencing COVID-19 severity13,19. Many genes highlighted in genetic studies on COVID-19 are implicated in key pathophysiological processes, including viral entry into cells, immunity, and inflammatory responses. Current genetic observations tend to a convergence of common and rare genetic variants that affect the interferon (INF) signaling pathways in patients with severe or critical COVID-1920. Although previous studies on COVID-19 were mostly conducted in Caucasian populations, Bulgarian ethnicity remains underrepresented in COVID-19 host genetic research.
In this study, we aimed to identify rare genetic variants in 444 Bulgarian patients with critical/severe, moderate and mild/asymptomatic COVID-19 using a whole exome sequencing (WES) approach, in order to establish an association between host genetic determinants and COVID-19 severity and outcome.
Methods
The ethics statement
The institutional board of the Ethics Committee of Medical University of Sofia (Bulgaria) approved all experiments including patient DNA (No 2187/29.06.20). Informed consent form was signed by all participants. The study adhered to the tenets of the Declaration of Helsinki for research involving human subjects.
Patients’ clinical data
This study was performed by collecting of a group of 444 unrelated patients with COVID-19 admitted to three of the biggest hospitals in Sofia, Bulgaria (including Military Medical Academy, University Multidisciplinary Hospital for Active Treatment and Emergency Medicine “N.I. Pirogov” and Acibadem City Clinic), from October 2020 to April 2022. All patients were of European ethnicity. The mean age is 56.8 ± 16.00 years (range 19–90): 181 females (41%; median age 60.8 ± 15.72 years) and 263 males (59%; median age 54.8 ± 16.17 years). SARS-CoV-2 infection was confirmed by positive PCR and/or serological test to detect the IgG and IgM antibodies. All participants were SARS-CoV-2 unvaccinated. The exclusion criteria were as follows: patients with terminal incurable diseases, immunodeficiency, long-term use of corticosteroids, pregnancy, alcoholism, drug addiction, and HIV.
Clinical information included primary demographic data, COVID-19 symptoms, co-morbidities, laboratory findings, treatments, COVID-19 complications and outcomes. An in-house maintained database was established for this project in order to create uniform medical record for each patient. Clinical data were extracted from medical records and patients were divided into three phenotypic groups according to the clinical severity and based on phenotype definitions of the World Health Organization (WHO): (0) Critical illness (individuals who have respiratory failure, septic shock, and/or multi-organ dysfunction, death); (1) Severe illness (severe pneumonia; adolescent or adult with clinical signs of pneumonia plus one of the following: respiratory rate > 30 breaths/min; severe respiratory distress; or SpO2 < 90% on room air); (2) Moderate illness (pneumonia; adolescent or adult with clinical signs of pneumonia but no signs of severe pneumonia, including SpO2 ≥ 90% on room air; (3) Mild illness (symptomatic patients who have any of the various signs and symptoms of COVID-19—fever, cough, sore throat, malaise, headache, muscle pain, nausea, vomiting, diarrhea, loss of taste and smell, but who do not have shortness of breath, dyspnea, or abnormal chest imaging); (4) Asymptomatic or pre-symptomatic infection (individuals who test positive for SARS-CoV-2 using a virologic test but who have no symptoms that are consistent with COVID-19) (Table 1). Severe or critical cases of COVID-19 have also been reported in individuals below 60 years of age who were previously healthy.
Whole-exome sequencing analysis
Genomic DNA of all subjects was isolated from peripheral blood using Chemagic DNA blood 10 k kit H1 on Chemagen Magnetic Separation Module (PerkinElmer®, Waltham, MA, USA) following the manufacturer’s instructions. Concentration of the genomic DNA was determined with the Qubit dsDNA BR Assay Kit on the Qubit 2.0 fluorimeter. DNA samples were fragmented, hybridized and captured using Illumina Exome Panel (Illumina, San Diego, CA, USA) according to manufacturer’s protocol. The libraries were tested for enrichment by qPCR, and the size distribution and concentration were determined using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Paired-end reads of 2 × 150 bp were generated per sample and WES with at least 97% coverage at 20×, as well as mean coverage uniformity at 94.9% (range 92–96%, SD: 0.96), was performed using the Illumina NovaSeq 6000 System (Illumina)12.
For WES analysis we applied a locally maintained DRAGEN (Illumina) secondary analysis pipeline for mapping to the GRCh37/hg19 human genome single nucleotide variants calling and quality filtering. Annotations and variant filtering were performed using VarSeq software (version 2.2.1, Golden Helix, Inc.) and as we have described previously21.
Rare variants analysis
To search for rare variants involved in COVID-19 pathophysiology, we used a combined gene panel. Candidate genes were selected due to their involvement in the pathways that are essential for SARS-CoV-2 entry, type I IFN system, primary immune-deficiencies, and genes related to coagulation. Additional genes that were previously reported to be associated with COVID-19 severity and susceptibility panel published in PanelApp22 (by selecting only green-labelled genes) were also chosen for the analysis. Besides, cardiovascular functionally related genes were also included. Finally, we summarized available resources of known risk variants and genes already associated with COVID-19 and collected these genomic regions as well. Thus, a total of 1172 genes were included in the final panel (Suppl. Table S1).
Variants were filtered according to a minor allele frequency (MAF) < 1% in population databases [the Genome Aggregation Database (gnomAD), https://gnomad.broadinstitute.org/]. Variant calls were required to have at least 20 × coverage (depth of 20 mapped reads) and quality score of minimum 20 (implying an accuracy > 99%)12,23. For heterozygous genotypes, the alternative allele ratio (Variant Allele Frequency, VAF) was set between 0.20 and 0.75 following variant quality control in similar studies12 and our in-house established protocol for WES analysis.
Variants with presumably disruptive impact on the protein, including splice acceptor variants, splice donor variants, stop gained, frameshift variants, stop lost, and start lost variants; missense variants (their pathogenicity depends on an amino acid change and a protein domain affected) and synonymous variants (they can also be functional because they can disrupt transcription, splicing, co-translational folding, mRNA stability, and can modulate gene expression by affecting transcription and splicing regulatory factors in protein-coding regions) were used for consideration. Intronic and non-coding variants were excluded from the analysis.
Mutation disease database ClinVar (ncbi.nlm.nih.gov/clinvar/) was used to identify variants previously reported as pathogenic/likely pathogenic and those described as likely benign/benign variants were discarded. The impact of missense variants was assessed using six predictor tools, SIFT24, PolyPhen225, MutationTaster26, MutationAssessor27, FATHMM-XF28, and FATHMM MKL Coding29. Finally, potential pathogenicity of prioritized variants was assessed following ACMG criteria30, which classify variants into 5 categories (benign, likely benign, uncertain significance, likely pathogenic, and pathogenic).
Statistical analysis
The statistical comparisons to assess the significance of the differences were made between phenotypic group 1 (patients with severe and critical COVID-19) against phenotypic groups 2 and 3 (patients with moderate, mild and asymptomatic COVID-19) taken together. P values were calculated by chi square test with Yates correction for association analysis of COVID-19 severity with the presence of rare novel variants, gender, comorbidities such as cancer, hypertension, cardiovascular disease, diabetes, chronic lung disease, as well as chest CT findings and acute respiratory distress syndrome (ARDS). Two tailed Fisher exact test was applied to assess the significance of differences between individuals presenting various values of binary traits for which one of the expected values is below 5 (for chronic kidney disease, ICU, pneumonia, unique variants). One-way ANOVA test with correlation analysis using Spearman's Rank Correlation Coefficient was performed to determine the significance of differences between age and disease course. Logistic regression (LR) was applied to estimate the association of the presence and the type of ventilation with disease outcome. The association test for all variants in genes from clusters obtained by STRING analysis were performed by clumping all detected rare pathogenic and likely pathogenic variants in the two severity groups (phenotypic group 1 versus phenotypic groups 2 and 3) and then compared using two tailed Fisher exact test with OR estimation. A statistical significance was considered at a p value < 0.05.
Principal component analysis
Since some of the results may be population-specific, we performed principal component analysis (PCA) of our 444 cohort’s population structure by projecting it onto 1000 Genomes (Phase 3, 2010/08/04 release date) principal component space built using the intersection of variants between the two datasets. The joint analysis with the 1000 Genomes dataset was conducted using PLINK231. The visualization was conducted using the R Statistical Software 4.2.3 (2023-03-15, http://www.r-project.org).
Quantile–Quantile (QQ) plots comparing all and rare variants quantiles representing the observations and their distribution with quantiles corresponding to the theoretical normal distribution.
Identification of pathogenic variants
From all detected rare variants we first filtered pathogenic and likely pathogenic variants according to ClinVar. All remaining rare variants such as variants of unknown significance (VUS), variants with conflicting interpretation as well as novel changes (not reported in gnomAD or dbSNP), displaying strong evidence for being considered as pathogenic/likely pathogenic following ACMG criteria, were also subjected to further analyses.
Functional network analysis on proteins of genes with detected pathogenic/likely pathogenic variants
Genes carrying variants that were previously described as pathogenic/likely pathogenic in ClinVar or displayed strong evidence for being considered as pathogenic/likely pathogenic following ACMG criteria were submitted to the STRING (version 12.0)32. STRING database integrates known and predicted interaction data across multiple organisms and collects data derived from different sources including gene co‐expression analyses, automated text‐mining and computational inference based on gene orthology.
Interactions with a STRING confidence score ≥ 0.4 as the threshold were downloaded as a file (.tsv) in short tabular text output format from the Exports tab. Cytoscape33 (version 3.10.1) was utilized for visualization. Clusters were defined as subgraphs with any two nodes (genes) connected to each other by edges (representing protein–protein association), and not connected to other nodes in the graph, these are normally called network components and the most extreme version of a cluster. We applied BINGO34 Cytoscape app for the enrichment analysis extracting over-represented Gene Ontology (GO, https://geneontology.org/) biological processes terms comparing their annotation in every cluster to the rest of the network including genes not grouped in clusters. In the network representation, the STRING combined score, which represents the interaction confidence, is used to characterize edges between genes.
Rare variants (MAF < 0.01) were subjected to burden analysis with covariates (age, sex and comorbidities) using the RVtest suite35. Several per-gene burden tests were chosen to compare their outcomes, including CMC36, CMCWald37, FP (“SCORE-Seq,” n.d.), and Zeggini38. Where applicable, multiple-testing correction was performed on the obtained p-values using four different methods (Bonferroni, Holm, Benjamini and Hochberg, Benjamini and Yekutieli). All gene burden methods were run for each gene from the human genome, but particular focus was placed on the currently used panel, containing genes of interest or medical relevance. Obtained p-values were adjusted for multiple testing using the four different methods.
Results
Clinical data of the studied cohort
A total of 444 COVID-19 unrelated patients (263 males and 181 females) admitted to the biggest hospitals in Sofia, Bulgaria, from October 2020 to April 2022, were included in the present study. They were divided into three qualitative phenotypic groups depending on the clinical severity: (1) patients with severe and critical course (181 cases; 41%); (2) individuals having moderate disease (129 cases; 29%); and (3) all cases who test positive for SARS-CoV-2 but who have some mild or no symptoms consistent with COVID-19 (134 cases; 30%). Phenotype definitions are described in Methods. The most frequent co-morbidities were hypertension, malignant diseases, and diabetes mellitus (Table 1). Detailed clinical data of each patient are recorded in a local database and could be provided on request.
Principal component analysis (PCA) showed that our cohort (collected in Bulgaria), with the exception of one sample, clusters together with the European population (Fig. 1A). The quantile–quantile (Q–Q) plot showed deflation of the observed p values, which means that our sample size is insufficient to analyze and interpret the results at the level of individual genetic variants (Fig. 1B,C).

Exome data of the studied cohort. (A) Principal components analysis (PCA) with 1,000 Genomes Project. COVID-19 samples from our study (black) and 1000 Genomes samples are plotted together based on principal components from overlapping SNP data. Five super populations: EUR, European; ASN, Asian; AMR, Ad Mixed American; AFR, African. (B) Quantile–Quantile (QQ) plot of association results for all variants; (C) QQ plot of association results for rare variants.
Analysis of the infected patients showed that the sex is significantly associated with the disease severity. Males are more likely to develop severe or critical disease than females. Relative risk for severe disease in males is 1.32 and OR = 1.59 (p = 0.024, 95% CI 1.07–2.35). Within the infected subjects, patients with severe infections and those who died were significantly older than the remaining clinical groups (asymptomatic/mild infection and moderate infection) (p = 0.002, ANOVA) (Table 1). Correlation analysis also showed significant association between age and disease severity (Spearman correlation coefficient – 0.193 with two tailed p = 4.06 × 10−5).
Recurrent genetic variants
An expected observation of our study was the high prevalence of subjects carrying three rare variants in the primary structural component of airways mucus gene, MUC5AC. These include c.1993A > G (rs36195734, MAF 0.02%) found in 302 subjects (68% in total; 28% severely ill), c.2018C > G (rs200292517, MAF 0.001%) identified in 281 (63% in total; 25% severely ill), and c.1974C > A (rs74811639, MAF 0.09%) which was detected in 147 (33% in total; 13% severely ill) patients.
Identification of pathogenic variants in COVID-19 patients
Given multiple previous reports related to the impact of pathogenic genetic variants on the severity and outcome of COVID-1939, we set off to test such effect using our cohort of 444 patients with varying degree of severity of the disease and clinical outcomes.
Using WES data, we focused on genes that have been involved in viral infection sensitivity, host immune response, genes related to coagulation, genes related to cardiovascular function, and genes that were previously shown to be associated with COVID-19 outcomes. Finally, we collected available information in a specific panel of 1172 genes (Suppl. Table S1). After applying filter steps described above, 9237 rare variants remained in the gene panel for 444 patients; the mean number of variants per person was 20.8. A total of 519 (out of 9237) were variants classified as either pathogenic or likely pathogenic, located in 244 different genes. These variants were observed in a total of 296 patients (Suppl. Table S2).
Unique variants
Our dataset of rare genetic variants found in patients with different COVID-19 outcomes was analyzed for alterations with unique combination of position and allele. The number of these variants was 5854 and they were located in 1083 different genes. They were detected in 439 patients, each carrying between 8 and 26 variants. Distribution of variants by type, MAF and functional consequences is shown in Fig. 2. Missense variants predominated among variants with other functional consequences (87%), synonymous changes counted 5.4%, and only 2.8% were represented by frameshift and splicing variants; (Fig. 2a). Ultra rare variants with MAF < 0.01% were the most numerous (Fig. 2b).

Whole exome sequencing data. (a) Distribution of variants by type and functional consequences. (b) By allele frequency (AF) according to population databases.
Fifty eight percent of the identified unique variants (n = 3395) were located in 673 genes involved in immune response. Seventy eight percent (n = 4558) of these rare variants were detected in genes with an autosomal recessive (AR) inheritance mode (CFTR, IL17RC, CCDC103, OAS2, OAS3, THBS3, ELF5, FBRSL1, SLC22A31, NR1H2, TYK2, IL36RN, DNAH5, VPS13B, etc.); 19.3% (n = 1134) of the variants were found in genes with an autosomal dominant (AD) inheritance mode (such as ABCA7, NFKB1, PLCG2, RNASEL, TLR3, BRCA1/2, CHEK2, MUC5B, and others), 1.7% (n = 99) variants were identified in X-linked genes (among which TLR7, ACE2 and G6PD), and 63 variants (1%) were located in overlapping AR and AD genes. Differences between patients with severe versus non-severe COVID-19 were not observed in the proportions of unique variants (p = 1, Fisher exact test).
Novel variants
Approximately 900 (n = 916; 9.9%) of the identified 9237 rare variants were novel, unreported in gnomAD or dbSNP (as of April 2024). Of note, 47 of those mutations (5.2%) appeared in heterozygous state in more than one (2 or 3) unrelated individuals (Table 2). Recurring extremely rare variants included synonymous, missense and frameshift changes. They were identified predominantly in individuals with mild course of the disease, although no conclusion as to the effect of the variants could be drawn considering the small number of patients involved.
Within this analysis, 93 variants subsequently classified as either pathogenic or likely pathogenic according to the ACMG criteria were identified. Among them, 38 were found in the subgroup of patients (n = 181, 41%) who developed severe complications during SARS-CoV-2 infection or deceased. These variants were dispersed across 34 distinct genes which are described in Suppl. Table S3. Twenty nine unreported in gnomAD or dbSNP pathogenic/likely pathogenic variants observed in this study have been submitted to the ClinVar database with the accession numbers SCV005091978–SCV005092006.
The subgroups corresponding to patients with moderate (n = 129, 29%), and mild/asymptomatic infections (n = 134, 30%) carried 55 novel pathogenic/likely pathogenic variants in total, which were located in 51 genes. (Suppl. Table S3). Genes were mainly connected to pathways for respiratory, immune, cardiovascular, and neurological diseases, all of which have been linked to the wide range of symptoms seen in severe, non-severe and long-COVID patients. Statistical analysis did not identify differences in the proportion of carriers of novel pathogenic genetic variants between the phenotypic groups (p = 0.89, χ2).
These results indicate that the pathogenic allele carrier status does not directly influence the course of COVID-19 in our patient group.
Genetic variants associated with SARS-CoV-2 severity
In our study, we did not find any of the rare variants in ACE1, , IFNAR2, TYK2, , CD40, FCGR2A, CASP3, DPP9, TLR3, TLR4, TLR7, TLR8, and TLR9 previously associated with prognosis and susceptibility to COVID-19 infection40,41,42, but we have detected ultra-rare variants (MAF < 0.01%) in three genes related to immune response (MUC5AC, ABCA7, FLNA), previously linked to the COVID-19 host response to different degrees43.
A variant, ACE2-c.1189A > G (p.Asn397Asp), found in a 37-year-old female (19334R) was included in a recently reported predisposing ACE2-genetic background44. The p.Gln665Glu variant in IL17RC, which was not described in public databases (dbSNP and gnomAD), was detected in a 58-year-old male from our cohort (18810R) who was diagnosed with autoimmune rheumatologic disease and diabetes type 2 at the time of the SARS-CoV-2 infection (Suppl. Table S2). This patient also carried the variants p.Ala40Glu and p.Glu329Gln located in the genes NFKB1 and TMPRSS2, respectively, which by its action facilitate the SARS-CoV-2 entry45. The same case was heterozygous for a pathogenic variant p.His154Pro in the gene CCDC103 associated with pulmonary symptoms in long-COVID patients46.
Rare variants affecting interferon signaling pathways (UNC93B1, IRF9, TLR3, IFNA1, TICAM1, IRF3, and IRF7) were identified in 7 (7/444; 1.6%) patients below 60 years of age who were previously healthy.
Novel genetic variants (not reported in the dbSNP and gnomAD) were detected in patients with critical and severe COVID-19 (Table 2, Suppl. Table S3). Among them, variants which were classified as pathogenic and possibly pathogenic according to ACMG criteria, were found in IL10RB, CENPF, DGKE, FYCO1, TONSL, CIB1, HLA-A, DCDC1, VPS13B and BRCA2 (Suppl. Table S3). Two of the severe patients (18012S and 18057S) shared a splice variant, c.1447-1G > T, in the gene CENPF, which according to the prediction programs is very likely to affect the splicing (Suppl. Table S3). An increase in the levels of centromeric protein CENPF which is involved in the regulation of cell cycle is observed as a result of viral infection suggesting that this protein is important for the viral replication control47. Therefore, it is possible that pathogenic variants in CENPF might influence the course of COVID-19. The c.804 + 2delT variant found in the IL10RB gene in a severely infected patient 17955S (Suppl. Table S3) represents a splice variant which, according to the prediction algorithms would affect splicing. IL10RB, encoding an interleukin receptor subunit, is reported to be a key regulator of COVID-19 host susceptibility and severity. Elevated blood levels of IL10RB are associated with a poor prognosis for the course of SARS-CoV-2 infection48, as it was recorded in case 17955S. Newly found variants in FYCO1 were detected in 3 patients with severe course of COVID-19. Two of these variants, classified as likely pathogenic, were identified in heterozygous state in patients with severe pneumonia, 18565R and 18291R (Suppl. Table S3). Variants of uncertain significance localized in FYCO1 were found in all clinical patient subgroups except the asymptomatic carriers.
Despite the absence of a significant effect of the presence of rare pathogenic variants on COVID-19 illness, we found that severely infected patients from our group carried pathogenic variants in genes linked to Mendelian disorders with increased susceptibility to infectious diseases, auto-inflammation, auto-immunity, allergy or malignancies. Pathogenic variants in the F7 (p.Ala354Val and p.Arg283Trp) and F11 (p.Gln244Ter and p.Trp519Ter) coagulation-related genes were found in patients with severe COVID-19 (Suppl. Table S2). Four severely affected cases (three males and one female) were carriers of a pathogenic variant c.563C > T (p.Ser188Phe) in the G6PD gene, mutations in which are associated with glucose-6-phosphate dehydrogenase deficiency (Suppl. Table S2). Pathogenic variants p.Arg4496Ter and p.Arg3539His in DNAH5 were detected in two patients with a critical and severe course of COVID-19, respectively (Suppl. Table S2). Variants in genes involved in the formation of cilia and flagellum have been suggested to be relevant to the body's response to SARS-CoV-2 infection. One of the deceased patients in our group, clinically diagnosed with hypertension (18542R), was a carrier of a pathogenic LoF variant in the DNAH5 gene, p.Arg4496Ter (Suppl. Table S2).
Heterozygous pathogenic variants in LDLR, p.Ala399Thr and p.Ser286Arg, were observed in three patients with severe and one with critical course of COVID-19, respectively (Suppl. Table S2). Eight CF-causing variants were identified in 9 patients with severe COVID-19 (Suppl. Table S2). A variant, c.2758G > T (p.Val920Leu), located in the AR gene CFTR and found to be deleterious in cystic fibrosis patients49 was identified in heterozygosity in the severely infected patient 18059S. Two cases with severe infection were carriers of well-known CF-disease causing mutations: CFTR-c.2491G > T (p.Glu831Ter) located in coding exon 1550, and CFTR-c.328G > C (p.Asp110His) located in coding exon 4 of the gene51.
We also noticed that some patients with critical or severe progression carried known pathogenic variants in genes linked to AD diseases. Mutations were found in genes related to cardiovascular disease (SCN5A, LDLR, DSG2), DNA damage repair response (CHEK2), coagulation (PROC), primary immune disorder (TNFRSF1A, COL7A1, LZTR1, CASP10), hemoglobin subunit β (HBB), and other genes (COL9A3, MUC5B, CHRND), associated with severe COVID-19. These cases required intubation and developed severe complications during SARS-CoV-2 infection or some of them died. Clinical features of the subjects carrying the identified variants, including the COVID-19 disease course and co-morbidities, are summarized in Supplementary Table S4.
This subset included three individuals with variants in the DNA damage repair response Fanconi anemia (FA)-BRCA pathway gene, CHEK2, which has been previously implicated in the pathogenesis of severe COVID-1952. In patients 18118R and 19110R we identified known pathogenic variant CHEK2-c.433C > T (p.Arg145Trp) that has been reported 20 times in ClinVar in hereditary cancer-predisposing syndrome patients (Suppl. Table S4). The second mutation, CHEK2-c.715G > A (p.Glu239Lys), found in patient 17759R is associated with familial breast cancer and is classified as pathogenic, supported by clinical interpretations previously reported in ClinVar53. Nevertheless, none of the patients had any symptoms of malignant tumors, although the ECOG score was changed in case 17759R (Suppl. Table S4).
Functional network analysis
Functional network analysis of genes with pathogenic/likely pathogenic variants
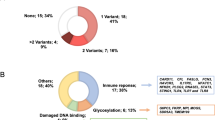
We further performed a network analysis, including 244 genes with pathogenic/likely pathogenic variants (Suppl. Table S2) to examine functional interactions between them. As a result, we identified 51 protein–protein interaction networks consisting of between 2 and 28 genes, and 9 singletons (Fig. 3). The majority of molecular functions, interactions and pathways are related to immune responses and DNA metabolism and repair, epithelial cilium movement and determination of left/right asymmetry, cellular response to oxidized low-density lipoprotein, regulation of lipid storage and lipid transport, complement activation; several interactions and pathways are related to the metabolic and cardiovascular systems, which could lead to multi-organ complications and dysfunction (Suppl. Table S5).

STRING network analysis demonstrating interaction between 244 risk proteins carrying 519 pathogenic/likely pathogenic variants. Legend: Network nodes represent proteins and edges represent protein–protein associations with a confidence score ≥ 0.4 as the threshold. Clusters are defined as subgraphs with any two nodes (genes) connected to each other by edges (representing protein-protein association), and not connected to other nodes in the graph. Networks containing less than three interacting proteins are not shown on the figure. risk protein, and networks 52–60 did not have any protein interaction with other human proteins. Cluster 2 containing genes involved in the epithelium cell movement is marked.
By comparing the clusters in terms of rare pathogenic/likely pathogenic changes carried by patients with severe/critical infections and the clinical groups of asymptomatic/mild and moderate cases we found one gene cluster that has an impact on severe outcomes of COVID-19. This main component contains 13 highly interconnected genes, ABCA7, CCDC103, CCNO, DNAH1, DNAH11, DNAH5, DNAH9, LVRN, NME8, STK36, SPAG1, GAS2L2, RAD51AP2 (Fig. 3, Table 3), related to epithelial cilium movement; mutations in these loci are associated with primary ciliary dyskinesia. The number of variants was 17, all in heterozygous state, and they were identified in 20 different patients (Table 3). Sixteen of these candidate variants were found in genes with an AR inheritance mode (such as CCDC103, CCNO, DNAH1, DNAH11, DNAH5, DNAH9, LVRN, NME8, STK36, SPAG1, GAS2L2, or RAD51AP2) and one variant was detected in gene with an AD inheritance mode (ABCA7). Statistical analysis reached significance in the assessment of all genes from the cluster among clinical groups. Results showed that patients carrying pathogenic or likely pathogenic variants in this gene cluster are more likely to develop critical or severe COVID-19 diseases with OR of 2.67 (95% CI 1.1–6.5), two tailed p = 0.028.
Variants DNAH11-c.11804C > T (p.Pro3935Leu) and CCDC103-c.461A > C (p.His154Pro) were found in three cases, and DNAH1-c.171delC (p.Lys58Serfs*25) was detected in two cases. The detected heterozygous frameshift variant in ABCA7, c.2871delC (p.Ser958Profs*34), was found in a 52-year-old male who was recorded with very severe pneumonia (18213R) and no co-morbidities. The p.Ser958Profs*34 variant in ABCA7 was not described in public databases (gnomAD) and was not found in other patients from our cohort (Table 3). According to the ACMG guidelines this change was classified as likely pathogenic. One of the deceased patients, clinically previously diagnosed with a hypertension (18542R), carried a pathogenic LoF variant in the DNAH5 gene (p.Arg4496Ter). Two cases with no pre-existing co-morbidities, one with moderate (18058S) and one with severe disease (19581R) carried two variants. Patient 18058S carried two likely pathogenic variants, c.11804C > T (p.Pro3935Leu) and c.607C > T (p.Arg203Ter), in DNAH11 and NME8, respectively. One pathogenic, CCDC103-c.461A > C (p.His154Pro), and one likely pathogenic change, LVRN-c.1856dupA (p.Asn619Lysfs*26), were found in a severely infected 72-year-old male (19581R).
Burden analysis
Rare variants (MAF < 0.01) were subjected to burden analysis with covariates (age, sex and comorbidities), but none of the genes from the selected panel reached statistical significance. A further analysis of the off-panel genes did not prove to be fruitful either, yielding no statistically significant results post correction (Suppl. Table S6).
Discussion
Since the emergence and rapid transmission of SARS-CoV-2, numerous scientific reports have searched for the association of host genetic variants with COVID-19, but no data are available for the Bulgarian population. In the present study, a group of 444 SARS-CoV-2 positive individuals were selected for a genetic study to search for the presence of rare pathogenic variants that may affect the course or outcome of COVID-19.
As expected, most of the detected rare patogenic variants (58%) were found in genes directly related to the immune response, as the gene panel used in this study was enriched in immune-related pathways as well as additional immune genes reported in previous studies55. Consistent with the association patterns, MUC5AC was the major transmembrane mucin of the respiratory tracts preventing microbial invasion in which rare variants were predominantly identified in severely infected patients. These data support the previously made conclusion that the prevalence, severity, and MUC5AC molecular basis of mucus accumulation is broadly similar in bronchioles and microcysts after SARS-CoV-2 infection56.
Rare pathogenic variants in single severely ill patients were found in the genes ACE2, IL17RC, NFKB1, TMPRSS2, CCDC103. Pathogenic variants in more than one patient were found in CFTR, CHEK2, SI, C9, DDX11, DNAH5, F11, F7, NPC1, G6PD, SCO2, LDLR, LIG4 and IL36RN.
Over the last years, a series of publications addressed the issue of hereditary predisposition to SARS-CoV-2 infection and severe disease. Analysis of genetic associations in cohorts of limited size, especially when no genome-wide genotypes are available, might also hinder the discovery of new susceptibility loci in underrepresented populations. The preliminary results from the COVID-19 Host Genetics Consortium include suggestive associations within the locus at chromosome 3p21.31 with the peak association signal covered a cluster of six potentially relevant to COVID-19 genes (SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, and XCR1)13. Single nucleotide polymorphisms or very rare variants in single genes have been reported as promising candidates as risk factors of severe COVID-19 in different populations14,15,17. In our study, we did not find any of the well-described rare variants in ACE1, IFNAR1, IFNAR2, TYK2, , CD40, FCGR2A, CASP3, DPP9, TLR2, TLR3, TLR4, TLR7, TLR8, and TLR9 associated with prognosis and susceptibility to COVID-19 infection40,41,42, but we have detected known mutations in genes related to cardiovascular disease (SCN5A, LDLR, DSG2), primary ciliary dyskinesia (DNAH5, DNAH, CCDC103), cystic fibrosis (CFTR), DNA damage repair response (CHEK2), coagulation (F7, F11, G6PD, PROC), primary immune disorder (TNFRSF1A, COL7A1, LZTR1, CASP10), hemoglobin subunit β (HBB), and other genes (COL9A3, MUC5B), associated with severe COVID-19. We have detected ultra-rare variants (MAF < 0.01%) in immune response-related genes, MUC5AC, ABCA7, FLNA, already associated with the host response to COVID-1943. As argued previously, a low replication rate might reflect both differences in study design and differences between populations14.
Additionally, we found 93 novel genetic variants classified as pathogenic or likely pathogenic in severely infected patients, 38 of whom developed massive complications during SARS-COV-2 infection or died. Among affected genes are some of the well-established candidates for severe disease, such as TNFRSF13B, MUC5B, ENTPD2, DPP9, SLC6A5, IL10RB, CENPF, FYCO1, RECQL4, VPS13B and BRCA2. Of note, 5.2% of these mutations were found in more than one patient. In light of these findings, it is important to consider the functions of these genes and their possible roles in COVID-19. Interestingly, two severely affected cases were carriers of novel likely pathogenic variants in the candidate susceptibility gene FYCO1, involved in the transport of autophagic vesicles. Pathak and co-authors summarized 27 genes playing a role in inflammation and coagulation pathways whose genetically predicted expression was associated with COVID-19 hospitalization. Among them, the authors found that FYCO1 was significantly associated with a severe progression of COVID-1957.
In our study, we observed significant association between rare genetic variants responsible for ciliary defects and severe progression of COVID-19, which contribute to the hypothesis that ciliary expressed genes are associated with severe disease. Early in the COVID-19 pandemic, chronic lung disease was considered as a risk factor for severe COVID-19 disease; people with pre-existing chronic health conditions are reportedly at high risk of catching the disease and of having a severe disease course58. Similar to other respiratory viruses, SARS-CoV-2 targets the ciliated cells of the respiratory epithelium and compromises mucociliary clearance, thereby facilitating spread to the lungs and paving the way for secondary infections59. Primary ciliary dyskinesia (PCD) is a multisystem genetic disease characterized by defects in motile cilia (ciliopathy), which plays an important role in several organ systems. Lung disease is a hallmark of PCD, given the essential role of cilia in airway surface defense. Studies have shown the higher risk of intensive care unit admission and mortality in COVID-19 patients with chronic obstructive pulmonary disease, cystic fibrosis, and PCD60. For this reason, people with PCD were strongly recommended to get vaccinated against SARS-CoV-261. Mutations in different dyneins and other realated to cilia formation proteins are associated with the AR disease, PCD. There is experimental evidence that SARS-CoV-2 infection induces states of ciliary and flagellar dysfunction62,63.
SARS-CoV-2 through its ORF10 protein targets the ubiquitin/proteasome pathway and subsequently leads to the loss of ciliary proteins, reduced viral particle clearance and allows for the continued spread of SARS-CoV-2 throughout the respiratory tract64. It also triggers formation of apically extended and highly branched microvilli that allow the viral export from the microvilli back into the mucus layer, supporting a model of virus dispersion throughout airway tissue via mucociliary transport65.
It is unclear how a heterozygous pathogenic mutation could contribute to the disease severity, but in our cohort we have observed 14 rare pathogenic mutations in genes related to cilia (DNAH1, DNAH5, DNAH9, DNAH11, CCDC103, STK36, RAD51AP2, ABCA7) in severe and critical cases, some of them found in more than one patient, compared to just 5 in moderate and 3 in mild cases (Table 3).
Mutations in other known rare disorders also may affect the severity of the COVID-19 infection. CFTR pathogenic mutations were found in 9 severely ill patients (2%) in our study and the finding is in line with the conclusions of Italian study of 874 patients, where the CF-carriers were more susceptible to the severe form of COVID-1966.
Overall, the severity of COVID-19 was high in our study cohort with 41% severe, critical or fatal disease, 29% with moderate disease and 30% asymptomatic or with mild infection. The next three factors may explain this. First, older age is one of the strongest predictors of severe COVID-19 as it was observed in our study and also reported before67; Eighty nine people (20%) were ≥ 70 years in our cohort, which may explain why we observed 6% of deaths. Secondly, 42% and 32% of the SARS-CoV-2 infections were collected during the time when the original strain (from January 2020 to December 2020) and Alpha (from January 2021 to May 2021) or Delta were dominant (from June 2021 to December 2021), respectively, which have been associated with more severe disease68. Thirdly, a combined not yet fully explored impact of common and rare exonic variants of host genetics on COVID-19 severity exists.
Risk factors such as age, co-morbidities, and environmental factors including socio-economic determinants of health, are known to have roles in disease severity. The effect of genetic factors ranging from those of rare, high-impact mutations that can modulate the difference between an asymptomatic individual (or developing mild symptoms) and life-threatening disease, to common genetic variants that only moderately affect symptom severity and susceptibility are still insufficiently understood.
Our study has several limitations. The main limitation is the limited sample size, when the sample is further divided into three subgroups based on severity. The association analysis of rare variants has insufficient power to identify candidate genes in gene-level tests to compare the results with those in the literature. Second, we have analyzed only the coding region of genes; thus, we could have missed a second pathogenic allele (deep intronic regions or CNVs) in monoallelic patients that could help us explain the COVID-19 outcome for genes with autosomal recessive inheritance. Another limitation is coming from the presumably complex and variable mechanisms determining the severity and outcome of COVID-19 disease. Risk factors affecting the course and outcome could be both genetic, related to the virus/host interaction, but could be also related to comorbidities, age, immune system status, as well as the choice of treatment. This presents further challenges in finding the best statistical methods to analyse such data.
However, our findings support the hypothesis that a dysregulation of interferon signaling pathways, in addition to the male sex and age, is associated with severe or critical COVID-1920. In the studied cohort, monogenic risk factors that affect the IFN-I signaling pathway showed a prevalence of 1.6% (expected between 1 and 5%) in young (< 60 years), patients with severe or critical COVID-19 without co-morbidities. Apart from known rare pathogenic variants in genes contributing to the disease severity, 93 novel variants in 38 severely affected patients were found in our cohort. Among the genes with rare pathogenic variants of specific interest is a cluster linked to the formation and functioning of the epithelial cilia, known to be used by SARS-CoV-2 both for the initial entry and further spread in our body following replication.
To our knowledge, this is the first large-scale genetic analysis of COVID-19 using whole exome sequencing of patients from the Bulgarian population.
Data availability
Novel (unreported in gnomAD or dbSNP) pathogenic/likely pathogenic variants observed in this study have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) and are publicly available online with the accessions SCV005091978–SCV005092006.
References
Ali, H. et al. Outcomes of COVID-19: Disparities by ethnicity. Infect. Genet. Evol. 87, 104639. https://doi.org/10.1016/j.meegid.2020.104639 (2021).
da Silva, S. J. R. et al. Two years into the COVID-19 pandemic: Lessons learned. ACS Infect. Dis. 8, 1758–1814. https://doi.org/10.1021/acsinfecdis.2c00204 (2022).
Cantalupo, S. et al. Regulatory noncoding and predicted pathogenic coding variants of CCR5 predispose to severe COVID-19. Int. J. Mol. Sci. 22, 5372. https://doi.org/10.3390/ijms22105372 (2021).
Hu, B., Guo, H., Zhou, P. & Shi, Z. L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 19, 141–154. https://doi.org/10.1038/s41579-020-00459-7 (2021).
Velavan, T. P. et al. Host genetic factors determining COVID-19 susceptibility and severity. EBioMedicine 72, 103629. https://doi.org/10.1016/j.ebiom.2021.103629 (2021).
Mitev, V. Comparison of treatment of COVID-19 with inhaled bromhexine, higher doses of colchicine and hymecromone with WHO-recommended paxlovid, molnupiravir, remdesivir, anti-IL-6 receptor antibodies and baricitinib. Pharmacia 70, 1177–1193 (2023).
Cappadona, C., Rimoldi, V., Paraboschi, E. M. & Asselta, R. Genetic susceptibility to severe COVID-19. Infect Genet Evol 110, 105426. https://doi.org/10.1016/j.meegid.2023.105426 (2023).
Lilov, A., Palaveev, K. & Mitev, V. High doses of colchicine act as “silver bullets” against severe COVID-19. Cureus 16, e54441. https://doi.org/10.7759/cureus.54441 (2024).
Nhung, V. P. et al. Host genetic risk factors associated with COVID-19 susceptibility and severity in Vietnamese. Genes 13, 1884 (2022).
Duman, M. et al. COVID-19 disease in presenting to the pediatric emergency department: A multicenter study of 8886 cases. Am. J. Emerg. Med. 59, 133–140. https://doi.org/10.1016/j.ajem.2022.06.015 (2022).
Kwok, K. O. et al. Editor’s choice: Influenza vaccine uptake, COVID-19 vaccination intention and vaccine hesitancy among nurses: A survey. Int. J. Nurs. Stud. 114, 103854. https://doi.org/10.1016/j.ijnurstu.2020.103854 (2021).
Butler-Laporte, G. et al. Exome-wide association study to identify rare variants influencing COVID-19 outcomes: Results from the host genetics initiative. PLoS Genet. 18, e1010367. https://doi.org/10.1371/journal.pgen.1010367 (2022).
Ellinghaus, D. et al. Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med. 383, 1522–1534. https://doi.org/10.1056/NEJMoa2020283 (2020).
Shcherbak, S. G. et al. Identification of genetic risk factors of severe COVID-19 using extensive phenotypic data: A proof-of-concept study in a cohort of Russian patients. Genes Basel 13, 534. https://doi.org/10.3390/genes13030534 (2022).
Tanimine, N. et al. Identification of aggravation-predicting gene polymorphisms in coronavirus disease 2019 patients using a candidate gene approach associated with multiple phase pathogenesis: A study in a Japanese city of 1 million people. Crit Care Explor 3, e0576. https://doi.org/10.1097/cce.0000000000000576 (2021).
Khadzhieva, M. B. et al. COVID-19 severity: Does the genetic landscape of rare variants matter?. Front. Genet. 14, 1152768. https://doi.org/10.3389/fgene.2023.1152768 (2023).
Santos-Rebouças, C. B. et al. Host genetic susceptibility underlying SARS-CoV-2-associated multisystem inflammatory syndrome in Brazilian children. Mol. Med. 28, 153. https://doi.org/10.1186/s10020-022-00583-5 (2022).
Pathak, G. A. et al. A first update on mapping the human genetic architecture of COVID-19. Nature 608, E1–E10. https://doi.org/10.1038/s41586-022-04826-7 (2022).
Mapping the human genetic architecture of COVID-19. Nature 600, 472–477. https://doi.org/10.1038/s41586-021-03767-x (2021).
van der Made, C. I., Netea, M. G., van der Veerdonk, F. L. & Hoischen, A. Clinical implications of host genetic variation and susceptibility to severe or critical COVID-19. Genome Med. 14, 96. https://doi.org/10.1186/s13073-022-01100-3 (2022).
Kamenarova, K. et al. Panel-based next-generation sequencing identifies novel mutations in Bulgarian patients with inherited retinal dystrophies. Mol. Genet. Genomic Med. 10, e1997. https://doi.org/10.1002/mgg3.1997 (2022).
Martin, A. R. et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 51, 1560–1565. https://doi.org/10.1038/s41588-019-0528-2 (2019).
Middha, S. et al. How well do whole exome sequencing results correlate with medical findings? A study of 89 Mayo Clinic Biobank samples. Front. Genet. 6, 244. https://doi.org/10.3389/fgene.2015.00244 (2015).
Sim, N. L. et al. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452-457. https://doi.org/10.1093/nar/gks539 (2012).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7–20. https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. https://doi.org/10.1038/nmeth0810-575 (2010).
Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 39, e118. https://doi.org/10.1093/nar/gkr407 (2011).
Rogers, M. F. et al. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 34, 511–513. https://doi.org/10.1093/bioinformatics/btx536 (2018).
Shihab, H. A. et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543. https://doi.org/10.1093/bioinformatics/btv009 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience https://doi.org/10.1186/s13742-015-0047-8 (2015).
Szklarczyk, D. et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646. https://doi.org/10.1093/nar/gkac1000 (2022).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Maere, S., Heymans, K. & Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. https://doi.org/10.1093/bioinformatics/bti551 (2005).
Zeggini, E. & Morris, A. Assessing rare variation in complex traits: Design and analysis of genetic studies (2015).
Li, B. & Leal, S. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321. https://doi.org/10.1016/j.ajhg.2008.06.024 (2008).
Lee, S., Abecasis, G. R., Boehnke, M. & Lin, X. Rare-variant association analysis: Study designs and statistical tests. Am. J. Hum. Genet. 95, 5–23. https://doi.org/10.1016/j.ajhg.2014.06.009 (2014).
Morris, A. P. & Zeggini, E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 34, 188–193. https://doi.org/10.1002/gepi.20450 (2010).
Shcherbak, S. G. et al. Identification of genetic risk factors of severe COVID-19 using extensive phenotypic data: A proof-of-concept study in a cohort of Russian patients. Genes 13, 534 (2022).
Mabrey, F. L., Morrell, E. D. & Wurfel, M. M. TLRs in COVID-19: How they drive immunopathology and the rationale for modulation. Innate Immun. 27, 503–513. https://doi.org/10.1177/17534259211051364 (2021).
Salihefendić, L. et al. Identification of human genetic variants modulating the course of COVID-19 infection with importance in other viral infections. Front. Genet. 14, 1240245. https://doi.org/10.3389/fgene.2023.1240245 (2023).
Matuozzo, D. et al. Rare predicted loss-of-function variants of type I IFN immunity genes are associated with life-threatening COVID-19. Genome Med. 15, 22. https://doi.org/10.1186/s13073-023-01173-8 (2023).
Mbarek, H. et al. Poking COVID-19: Insights on genomic constraints among immune-related genes between Qatari and Italian populations. Genes (Basel) 12, 1842. https://doi.org/10.3390/genes12111842 (2021).
Benetti, E. et al. ACE2 gene variants may underlie interindividual variability and susceptibility to COVID-19 in the Italian population. Eur. J. Hum. Genet. 28, 1602–1614. https://doi.org/10.1038/s41431-020-0691-z (2020).
Li, Y., Wang, C., Chang, Y., Chen, C. & Chen, Y. Differentially expressed FOXO1, NFE2L2 and NFKB1 mRNAs are associated with differentially regulated corona- and influenza viral receptor genes and Toll-like receptor pathway genes in human bronchial epithelial cells under hyperbaric oxygen exposure. Eur. Respir. J. 60, 168. https://doi.org/10.1183/13993003.congress-2022.168 (2022).
Sanisha, D. & Suresh, K. Long COVID: G protein-coupled receptors (GPCRs) responsible for persistent post-COVID symptoms. bioRxiv https://doi.org/10.1101/2022.12.12.520110 (2022).
Ghandikota, S., Sharma, M. & Jegga, A. G. Secondary analysis of transcriptomes of SARS-CoV-2 infection models to characterize COVID-19. Patterns (N Y) 2, 100247. https://doi.org/10.1016/j.patter.2021.100247 (2021).
Voloudakis, G. et al. (medRxiv, 2021).
Picci, L. et al. A 10-year large-scale cystic fibrosis carrier screening in the Italian population. J. Cyst. Fibros. 9, 29–35. https://doi.org/10.1016/j.jcf.2009.10.003 (2010).
Cutting, G. R. et al. A cluster of cystic fibrosis mutations in the first nucleotide-binding fold of the cystic fibrosis conductance regulator protein. Nature 346, 366–369. https://doi.org/10.1038/346366a0 (1990).
Dean, M. et al. Multiple mutations in highly conserved residues are found in mildly affected cystic fibrosis patients. Cell 61, 863–870. https://doi.org/10.1016/0092-8674(90)90196-l (1990).
Bolton, K. L. et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat. Commun. 12, 5975. https://doi.org/10.1038/s41467-021-26138-6 (2021).
Wu, X., Dong, X., Liu, W. & Chen, J. Characterization of CHEK2 mutations in prostate cancer. Hum. Mutat. 27, 742–747. https://doi.org/10.1002/humu.20321 (2006).
López-Rodríguez, R. et al. Presence of rare potential pathogenic variants in subjects under 65 years old with very severe or fatal COVID-19. Sci. Rep. 12, 10369. https://doi.org/10.1038/s41598-022-14035-x (2022).
Liu, Z. M., Yang, M. H., Yu, K., Lian, Z. X. & Deng, S. L. Toll-like receptor (TLRs) agonists and antagonists for COVID-19 treatments. Front. Pharmacol. 13, 989664. https://doi.org/10.3389/fphar.2022.989664 (2022).
Chatterjee, M., van Putten, J. P. M. & Strijbis, K. Defensive Properties of Mucin Glycoproteins during Respiratory Infections-Relevance for SARS-CoV-2. mBio 11, 7. https://doi.org/10.1128/mBio.02374-20 (2020).
Pathak, G. A. et al. Integrative genomic analyses identify susceptibility genes underlying COVID-19 hospitalization. Nat. Commun. 12, 4569. https://doi.org/10.1038/s41467-021-24824-z (2021).
Noor, Z. S., Cummings, A. L., Johnson, M. M., Spiegel, M. L. & Goldman, J. W. Targeted therapy for non-small cell lung cancer. Semin. Respir. Crit. Care Med. 41, 409–434. https://doi.org/10.1055/s-0039-1700994 (2020).
Schreiner, T. et al. SARS-CoV-2 infection dysregulates cilia and basal cell homeostasis in the respiratory epithelium of hamsters. Int. J. Mol. Sci. 23, 5124. https://doi.org/10.3390/ijms23095124 (2022).
Gerayeli, F. V. et al. COPD and the risk of poor outcomes in COVID-19: A systematic review and meta-analysis. EClinicalMedicine 33, 100789. https://doi.org/10.1016/j.eclinm.2021.100789 (2021).
Caminati, M. et al. COVID-19 vaccination in patients with severe asthma on biologic treatment: Safety, tolerability, and impact on disease control. Vaccines (Basel) 9, 853. https://doi.org/10.3390/vaccines9080853 (2021).
Bermejo-Valdés, A. J., Padrón-González, A. A. & Archer-Jiménez, J. in Preprints (Preprints, 2020).
Robinot, R. et al. SARS-CoV-2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nat. Commun. 12, 4354. https://doi.org/10.1038/s41467-021-24521-x (2021).
Wang, L. et al. SARS-CoV-2 ORF10 impairs cilia by enhancing CUL2ZYG11B activity. J. Cell Biol. 221, e202108015. https://doi.org/10.1083/jcb.202108015 (2022).
Wu, C. T. et al. SARS-CoV-2 replication in airway epithelia requires motile cilia and microvillar reprogramming. Cell 186, 112-130.e120. https://doi.org/10.1016/j.cell.2022.11.030 (2023).
Baldassarri, M. et al. Severe COVID-19 in hospitalized carriers of single CFTR pathogenic variants. J. Pers. Med. 11, 558. https://doi.org/10.3390/jpm11060558 (2021).
Noor, F. M. & Islam, M. M. Prevalence and associated risk factors of mortality among COVID-19 patients: A meta-analysis. J. Commun. Health 45, 1270–1282. https://doi.org/10.1007/s10900-020-00920-x (2020).
Earnest, R. et al. Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England, USA. Cell Rep. Med. 3, 100583. https://doi.org/10.1016/j.xcrm.2022.100583 (2022).
Acknowledgements
We are grateful to all patients who participated in the study. We thank the staff (physicians and nurses) of the three Sofia hospitals, for the medical care and treatment of the patients. Furthermore, we thank our families for providing us with the time and support needed to write this survey in a timely manner.
Funding
This study is supported by the National Science Fund of Bulgarian Ministry of Education and Science under Grant number КP-06-DK1/8/2021 and Infrastructure Grants D01/ 285/2019; D01-395/2020; D01-302/2021 by the Ministry of Education and Science.
Author information
Authors and Affiliations
Contributions
K.K., D.K.-Y., I.D., V.M., and R.K. contributed to the design of the study. M.B., R.A., P.A., M.K., R.A., A.Z., R.D., I.I., D.P., V.K., A.P., D.L.-N., K.P., G.P., I.P., and N.G. provided samples and clinical data. M.B., M.G, and I.I. reviewed the clinical data and classify patients in severity categories. K.M. performed the N.G.S. D.K.-Y., M.G., V.P., and D.G. performed the bioinformatic and statistical analysis. K.K., D.K.-Y., O.B., D.P., R.B., N.I., and R.K. contributed to the interpretation and analysis of the genetic data. All authors contributed to the review and approval of the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kamenarova, K., Kachakova-Yordanova, D., Baymakova, M. et al. Rare host variants in ciliary expressed genes contribute to COVID-19 severity in Bulgarian patients. Sci Rep 14, 19487 (2024). https://doi.org/10.1038/s41598-024-70514-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-70514-3
Keywords
This article is cited by
-
Unraveling the protective genetic architecture of COVID-19 in the Brazilian Amazon
Scientific Reports (2024)
