
Abstract
Gliomas are the most common tumor in the central nervous system in adults, with glioblastoma (GBM) representing the most malignant form, while low-grade glioma (LGG) is a less severe. The prognosis for glioma remains poor even after various treatments, such as chemotherapy and immunotherapy. Cuproptosis is a newly defined form of programmed cell death, distinct from ferroptosis and apoptosis, primarily caused by the accumulation of the copper within cells. In this study, we compared the difference between the expression of cuproptosis-related genes in GBM and LGG, respectively, and conducted further analysis on the enrichment pathways of the exclusive expressed cuproptosis-related mRNAs in GBM and LGG. We established two prediction models for survival status using xgboost and random forest algorithms and applied the ROSE algorithm to balance the dataset to improve model performance.
Similar content being viewed by others

Introduction
Glioma is one of the most common central nervous system tumors worldwide, encompassing both the malignant glioblastoma (GBM) and low-grade gliomas (LGG)1,2. LGG is classified as a World Health Organization (WHO) grade II/III tumor, while GBM is classified as grade IV3. In 2021, the WHO reclassified over 15 diffuse gliomas into three types: (1) astrocytoma (WHO grade II-IV); (2) Oligodendroglioma (WHO grade II and III); and (3) glioblastoma (WHO grade IV)4. Since GBM is a more severe tumor comparing to LGG, the median survival time for GBM ranges from 14 to 17 months5,6. Meanwhile, the survival rate for patients with LGG is significantly higher than for those with GBM7,8. Due to the invasive nature of glioma, LGG is likely to progress to GBM within months. Another characteristic of glioma is its variability; different IDH mutation and the combination of 1p/19q co-deletion also affect the survival status of LGG9,10.
Cell death is a complex and essential process with various types, including apoptosis, necrosis, autophagy, ferroptosis, cuproptosis, pyroptosis and necroptosis11,12,13,14. Cuproptosis, a newly defined form of programmed cell death, occurs when excessive copper induces cell death15,16. Copper is a fundamental element in maintaining enzyme function, but an excessive amount of copper can trigger cell death17,18. Research also indicates that cells undergoing cuproptosis are closely related to mitochondrial respiration19. The standard treatments for glioma, such as surgery and radiation, are not entirely effective, making the search for new treatments essential to improving the survival of glioma patients20. Mudassar et al. presented evidence that an anti-parasitic drug could improve the effectiveness of radiotherapy in high-grade gliomas by targeting mitochondrial metabolism, which aligns with the function of cuproptosis21. Several studies have identified genes associated with copper-induced cell death, laying the foundation for cuproptosis-related gene research22,23.
Noncoding RNAs include microRNAs (miRNAs), long non-coding RNAs (lncRNAs), PIWI-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), and circular RNAs (circRNAs)24,25. LncRNAs are a class of noncoding RNAs longer than 200 nucleotides and do not encode proteins. LncRNA research has recently become a spotlight topic, with numerous studies demonstrating that lncRNAs play an essential role in cell cycles26, gene transcription regulation and post-transcriptional processes, and more27. Dysregulation of lncRNAs, whether under- or over-expressed, may contribute to the onset of various diseases. Several studies have developed prognostic models based on lncRNAs and gliomas to identify biomarkers that could potentially improve survival rate of gliomas23,28. Additionally, mRNAs are a class of nuclide acids that carry protein-coding information, essential to the functioning of living organisms. As research on mRNA and cancer increaes, studies have shown that mRNA could serve as a biomarker for improving the prognosis of various cancers29,30,31.
In the present study, we explored the role of cuproptosis-related lncRNAs and mRNAs in predicting the survival status of gliomas. We aimed to build a predictive model using several lncRNAs or mRNAs to predict the survival of LGG and GBM patients, employing random forest and Xgboost algorithms. We hope our model would help improve the diagnosis and treatment of glioma. Moreover, the difference in the role of cuproptosis between LGG and GBM from a bioinformatics aspect has not been well studied. Therefore, another aspect of our study is to identify differences between LGG and GBM when introducing cuproptosis-related genes.
Results
The relationship and correlations among cuproptosis genes in GBM and LGG
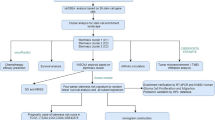
The flowchart of our study is illustrated in Fig. 1. Among the identified cuproptosis genes, we observed that the gene ATP7A had a relatively high correlation with most of the other cuproptosis genes (Fig. 2A). In Fig. 2A,B, the color represents the direction of correlations, and the numbers indicate the strength of these correlations. Moreover, only correlations with a p-value smaller than 0.05 were displayed in the heatmap. Among the 17 cuproptosis genes, the highest correlation was observed between LIAS and LIPT1. Conversely, the correlation between CDKN2A and other genes was nearly 0, except for ATP7A, PDHA1, and DBT. In Fig. 2B, the strongest correlation was between MTF1 and DBT. Overall, the correlations among cuproptosis in GBM are greater than in LGG, and all correlations in GBM were positive, whereas some correlations among cuproptosis genes in LGG were negative. In Fig. 2C,D, the expression levels of cuproptosis genes in GBM and LGG are shown in the form of heatmap. In the heatmap for GBM (Fig. 2C), the expression levels appeared mostly in green, indicating lower expression after standardization. We were able to discern the expression differences between GBM and LGG in both heatmaps (Fig. 2C,D).

Flowchart illustrating the study design.

The relationship and correlations among cuproptosis genes in GBM and LGG. (A) Correlation heatmap of cuproptosis genes in GBM. (B) Correlation heatmap of cuproptosis genes in LGG. (C) Heatmap of expression level of cuproptosis genes clustered by clinical information. (D) Heatmap of expression level of cuproptosis genes clustered by different types of gliomas (LGG, GBM). (E) Boxplot comparing the expression levels of cuproptosis genes between GBM and LGG. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (F) Protein–Protein Interaction (PPI) network of cuproptosis genes in GBM.
In the boxplot comparing the differences in the expression level of cuproptosis genes between GBM and LGG, we observed that only five out of 17 genes did not show significantly differences between GBM and LGG (Fig. 2E). Moreover, most cuproptosis genes had higher expression in LGG compared to GBM. The Protein–Protein interaction (PPI) network of cuproptosis genes is shown in Fig. 2F. We performed consensus clustering analysis on both GBM and LGG, but only the clustering in GBM yielded meaningful results. The analysis classified the genes in GBM into two clusters, although the classification based on the PCA was not ideal (Supplement Fig. 1).
Functional analysis of exclusive genes in GBM and LGG
The results of the GO enrichment analysis of genes exclusive to GBM and LGG are shown in Fig. 3A,B, respectively. The bubble plots indicate that the biological process, cellular component, and molecular functions were different for the exclusive genes in the two types of gliomas. We also performed KEGG enrichment analysis on the exclusive genes in GBM and LGG, separately. We discovered that a considerable number of genes in GBM were enriched in pathways related to neurological diseases, such as Huntington disease and Parkinson disease. On the other hand, the genes in LGG were enriched in only one pathway, insulin secretion. This finding in LGG mat serve as a distinguishing factor for patients who have LGG and have not yet progressed to GBM.

Functional analysis of genes exclusive to GBM and LGG. (A) Bubble plot of GO analysis for exclusively expressed mRNAs in GBM. (B) Bubble plot of GO analysis for exclusively expressed mRNAs in LGG.
The KEGG pathways in GBM were also enriched for various neurological diseases, consistent with the GO analysis (Fig. 4A). In contrast, insulin secretion was the only enrichment pathway identified in LGG (Fig. 4B). Combined with the patients’ clinical information, we were able to distinguish differences in survival probability between GBM and LGG. In this study, we were particularly interested in how the genes exclusively to GBM and in LGG differ in term of survival and prognostic values. The Venn diagrams illustrate the number of cuproptosis-related mRNAs and lncRNAs in GBM and LGG, respectively (Fig. 4C,D). The selection criteria are described in detail in the Method section. There were 333 lncRNAs shared between cuproptosis-related lncRNAs in GBM and LGG. Moreover, the number of exclusively expressed cuproptosis-related lncRNAs in GBM was almost 10 times greater than in LGG. This trend was also observed for cuproptosis-related mRNAs in GBM and LGG. One hypothesis is that cuproptosis genes are more highly expressed more in GBM, where the disease is more severe than in LGG. As shown in Fig. 4E, we observed that the survival probability of patients with GBM was significantly lower than that of patients with LGG (p < 0.0001), which is expected given that GBM is a more aggressive tumor than LGG.

KEGG pathway analysis of exclusively expressed mRNAs in GBM and LGG. (A) Bubble plot of KEGG pathway analysis for mRNAs in GBM. (B) Bubble plot of KEGG pathway analysis for mRNAs in LGG. (C) Venn Diagram showing genes filtered based on the correlation between cuproptosis genes and lncRNAs in gliomas. (D) Venn Diagram showing genes filtered based on the correlation between cuproptosis genes and mRNAs in gliomas. (E) Kaplan–Meier survival curves for GBM and LGG.
Feature selection using the elastic net algorithm
To select appropriate genes for further analysis, we employed the elastic net method. The accuracy of elastic net classification for mRNA from GBM and LGG is demonstrated in Fig. 5A,B. The accuracy fluctuated with different regularization parameters during repeated cross-validation (Fig. 5C,D). The best performance occurred when the parameter was 0.05 for GBM and 0.2 for LGG using the elastic net method, with accuracy being slightly higher in GBM than in LGG. The bar plots demonstrated the variable importance in the two finalized models. By combining survival information, we were able to select genes related to survival. Unfortunately, we were not able to identify any survival-related mRNAs from GBM, although three genes in LGG were survival-related, including CALM3, SMIM17, and NCS1 (Fig. 5E,F). We also performed the same analysis to lncRNAs in both GBM and LGG. We observed that the best-performing regularization parameter was 0.2 for both GBM and LGG, with accuracy being better in GBM than in LGG, which was consistent with the findings for mRNAs (Fig. 6A,B). The importance of the selected features was demonstrated, showing the variables in descending order of importance (Fig. 6C,D). Unfortunately, we were unable to identify any survival-related lncRNAs in either GBM or LGG (Fig. 6E,F).

Feature selection of cuproptosis-related mRNAs using elastic net. (A, B) Accuracy of elastic net classification for mRNAs in GBM and LGG. (C, D) Variable importance of the selected features from elastic net for GBM and LGG, with higher importance at the top. (E, F) Forest plots of hazard ratios of selected genes in GBM and LGG, with red dots indicating HR > 1 and green dots indicating HR < 1.

Feature selection of cuproptosis-related lncRNAs using elastic net. (A, B) Accuracy of elastic net classification for lncRNAs in GBM and LGG. (C, D) Variable importance of selected feature from elastic net for GBM and LGG, with higher importance at the top. (E, F) Forest plots of hazard ratios for selected genes in GBM and LGG, with red dots indicating HR > 1 and green dots indicating HR < 1.
Feature selection and model prediction using the random forest algorithm
Due to the nature of our data, the survival rates for GBM and LGG were distinctly different. In GBM, the death rate was over 81% death, whereas in LGG it was only 24%. Therefore, we used the ROSE algorithm to over-sample the minority class to achieve a more balanced dataset. We then used the random forest algorithm to predict survival status on both unbalanced and balanced data, comparing the difference before and after the data adjustment. In the random forest analysis, the importance of each features was measured using Mean Decreasing Accuracy and Mean Decrease Gini methods for mRNAs and lncRNAs in GBM, respectively (Fig. 7A,B). The AUC for cuproptosis-related lncRNAs and mRNAs was 1 in the training set, which was surprisingly high. However, the AUC for both groups in the test set was only 0.52 and 0.51 (Fig. 7C,D), which was just barely over 0.5. After applying the ROSE algorithm, the AUC for cuproptosis-related mRNAs decreased to 0.85 in the training set, while the AUC in the test set increased by 0.01 compare to earlier dataset (Fig. 7E). The results in the AUC of cuproptosis-related lncRNAs were slightly better compared to mRNAs, with the AUC in the training set decreasing only to 0.96, while the AUC in the test increased by 0.02 (Fig. 7D,F). We also performed the same analysis in LGG, and the results were similar to those for GBM, with balanced data constructed through ROSE. The AUC improved slightly in both mRNAs and lncRNAs compared to unbalanced data (Supplement Fig. 2).

Variable importance and ROC curves in random forest analysis. (A) Variable importance bubble plot for mRNAs in the GBM training set using random forest. (B) Variable importance bubble plot for lncRNAs in the GBM training set using random forest. (C, D) ROC curves for the training and test sets before ROSE data adjustment, for mRNA and lncRNA, respectively. (E, F) ROC curves for the training and test sets after ROSE data adjustment, for mRNA and lncRNA, respectively.
Discussion
Gliomas are heterogeneous, particularly LGG, as it can be found in various locations in the brain, such as the brainstem and spinal cord. Therefore, the characteristics of the tumor can vary, including the type of treatment and survival rate32,33. Starting in 2016, the WHO classified grade II-IV diffuse gliomas into two groups based on the mutations in the isocitrate dehydrogenase (IDH1/2). Glioblastoma can be further classified based on the molecular classification (IDH wildtype and IDH mutant)34,35,36,37. LGG has the potential to progress to GBM within 3 to 5 years, and the survival rate dramatically decreases9,38. This was also validated in the data in this study, showing that patients with LGG had significantly better survival rates than those with GBM.
The most common treatment for glioma patients is still surgery; however, the survival after surgery is not ideal39. Therefore, identifying underlying biomarkers for gliomas to improve the survival rate is urgently needed. Multiple studies have indicated that cancer immunotherapy could improve the survival of patients with gliomas and other cancers40,41,42. One study developed a score based on the survival and clinical information of cuproptosis-related genes, indicating that the score is negatively correlated with survival time in gliomas, regardless of the stages43. It has also been discovered that promoting the oxidation of proteins in glioma cells inhibits these cells by the copper ions. A cuproptosis-related gene signature model was constructed, identifying five genes as independent prognostic predictors in LGG patients44.
In this study, we filtered out the lncRNAs and mRNAs that are cuproptosis-related based on a series of selection criteria. We observed that most cuproptosis gene expression in LGG are higher than in GBM. It is reasonable to speculate that the expression level of copper in cells might lead to different glioma grade and thus affect survival. Our results are consistent with a recent study that suggested lower expression of FDX1 leads to longer survival, whereas higher expression of CDKN2A is associated with longer survival45. A six cuproptosis-related genes signature was found to support the idea that patients in the high-risk group have worse survival outcomes, and significantly differences were also found between the immune cells in the two risk groups46. Wang et al. identified cuproptosis-related lncRNAs for immunotherapy and prognosis in glioma47. Two other studies revealed a signature of cuproptosis-related lncRNAs that predicts prognosis in glioma48,49. Wen et al. identified a cuproptosis-related lncRNA signature for prognostic value and therapeutic prediction for LGG. Five cuproptosis-related lncRNAs contributed to predicting survival of LGG50. Yan et al. reported a cuproptosis-associated lncRNA signature for forecasting prognosis, chemotherapy efficacy, and response to immune checkpoint blockade therapy in LGG51. Xu et al. also analyzed prognosis and immune response of a cuproptosis-related lncRNA signature in LGG52.
We identified lncRNA ZFAS1 (Zinc finger antisense 1) as a cuproptosis-related lncRNA in GBM in this study. Indeed, lncRNA ZFAS1 has been reported to be involved in glioma development. Zhang et al. reported that lncRNA ZFAS1 facilitated glioma development via regulation of miR-1271-5p and HK2, leading to increased proliferation, invasion and migration53. Li et al. found that lncRNA ZFAS1 enhanced the progression of glioma by targeting the miR-150-5p/PLP2 axis54. One group showed that ZFAS1 activated the Notch signaling pathway and promoted glioma progression55. Another group identified that overexpression of ZFAS1 induced EMT and was associated with poor prognosis in glioma56. In the functional enrichment analysis, we found that the only KEGG pathway identified for LGG was insulin secretion, which did not appear in the pathways for GBM. A study has indicated that patients with high insulin intake were more likely to develop gliomas when controlling for other confounders57. However, this study did not specify the grade of glioma the patients experienced. We were surprised to see the connection between insulin and LGG in the KEGG analysis. Nevertheless, we recognized a few shortcomings in our study. First, we lack sufficient clinical information to perform more detailed analyses, especially with no record on the grade of GBM. Secondly, since we were specifically looked for lncRNAs and mRNAs that were exclusively expressed in GBM and LGG, we lost some information carried by the shared genes. These shared genes had a higher correlation with cuproptosis genes than the exclusive genes, which might have prevented us from identifying a few survival-related genes, leading to the result that none of the genes in our study were significantly related to survival. Moreover, we do not have enough data to support our analysis, particularly as the sample size for GBM is almost one-quarter of that for LGG. More importantly, when applying the unbalanced dataset algorithm to GBM, the sample size of GBM was further reduced. Furthermore, the genes that showed relevance with survival did not be approved with experimental results. Thus, further study could be conducted based on our work, and with more data, we believe the results of our study could be improved.
In conclusion, we found that a majority of cuproptosis genes were highly expressed in LGG compared to GBM. Additionally, we identified the insulin secretion pathway as exclusively enriched in LGG, which does not appear in the KEGG pathway in GBM. Using elastic net and random forest to select features based on patient survival status, none of the features selected by elastic net was survival-related. With the ROSE algorithm, the balanced dataset provided slightly better predictions than the original data using the random forest algorithm for both GBM and LGG. According to the literature, no studies have explored cuproptosis-related circRNAs in gliomas. It is necessary to investigate cuproptosis-related circRNAs in gliomas. The integration of multi-omics data, including genomics, transcriptomics, proteomics, metabolomics and epigenomics, should be performed to provide a more comprehensive understanding of the development and progression of glioma. The application of single-cell sequencing and spatial omics techniques will allow for the analysis of cellular heterogeneity and spatial organization within glioma tissues. Additionally, our bioinformatics predictions and analyses should be validated through in vitro and in vivo experiments.
Methods
Data collection
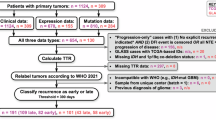
In this study, we obtained RNA-seq data of glioma patients through the TCGA database, which includes 169 GBM patients and 532 LGG patients. We also extracted corresponding clinical information (survival time, survival status, gender, age) of those patients from the TCGA database (Tables 1 and 2). All 701 samples collected from TCGA were tumor tissues, so we harvested 1132 normal tissues from the GTEx database for comparison. Additionally, we gathered 17 cuproptosis-related genes from several literatures18,22,23. All data from TCGA and GTEx were transformed into log2(x + 1) for further analysis. To building the prediction model, we split the GBM and LGG patients into 60% training and 40% testing sets.
Cuproptosis genes correlation
To investigate the relationship among cuproptosis genes in GBM and LGG, we applied Pearson correlation to the 17 cuproptosis-related genes and presented the results in a heatmap. We also compared the differences in cuproptosis gene expressions between GBM and LGG using a boxplot, with genes on the X-axis and expression level on the Y-axis. A heatmap showing the differences in gene expression was also generated.
We were particularly interested in identifying lncRNAs and mRNAs correlated with cuproptosis genes. Firstly, we calculated the Pearson correlation between lncRNAs and cuproptosis, selecting only lncRNAs with an absolute correlation value greater than 0.5 and p < 0.05. Similarly, we selected cuproptosis-related mRNAs with an absolute correlation value greater than 0.7 and p < 0.001 from both GBM and LGG. Since the number of mRNAs is much greater than that of lncRNAs, the selection criterion for mRNAs was more stringent. We then created Venn diagrams of the selected lncRNAs and mRNAs in GBM and LGG to filter out the common genes shared between the two stages of gliomas.
GO/KEGG
We filtered out the exclusively expressed lncRNAs and mRNAs by removing the shared genes between GBM and LGG. For the distinct pathways in non-overlapping cuproptosis-related lncRNAs and mRNAs in GBM and LGG, we performed GO and KEGG enrichment analysis58,59,60 with the “clusterProfiler” and “limma” packages in R Studio, with the following criteria: p adjust < 0.05 and |\({\text{log}}_{2}FC\)|≥ 1.
Survival analysis
We were interested in exploring the differences between GBM and LGG, as these are two stages of gliomas, and GBM is associated with poorer survival. The Kaplan–Meier curve comparing the two stages of glioma aligns with this observation. The clinical information for GBM was summarized in Table 1, which shows that, compared to LGG, GBM lacks information on the tumor grade.
Consensus clustering
Given the lack of information on GBM grade, we aimed to identify subtypes of GBM patients through consensus clustering analysis. The analysis was conducted using the “limma” package in R. The number of subtypes ranged from 2 to 8, and survival analysis was performed on the best-performed number of subtypes.
Feature selection
To save computational power, we constrained the number of cuproptosis-related mRNAs and lncRNAs to the top 100 cuproptosis-correlated genes. Since the survival rates for GBM and LGG are distinctly different, predicting a patient’s survival status is important for clinical purpose. Furthermore, identifying several biomarkers to aid in prediction is fundamental. The common feature selection method for prognostic genes is Lasso regression; however, it has several shortcomings, particularly when the dimension is high and variables are high correlation. In this case, Lasso regression may not be suitable for selecting multiple genes that are highly correlated, as it may conserver only one or two genes and lose valuable information in the process. Another limitation is that when the number of genes (p) exceeds the number of patients n, we can only select n genes.
In this study, we proposed using random Lasso and elastic net to address the limitations mentioned above. The random Lasso method generates thousands of datasets through bootstrap, which contributes to variable selection. The parameter is also more flexible than regular Lasso, as the number of variables selected in each bootstrap can cary and can be as large as the number of patients. Compared to Lasso regression, elastic net adds an additional penalty from Ridge regression, making it more restrictive than regular Lasso regression.
Unbalanced dataset
Since GBM and LGG are essentially different stages of gliomas, and numerous studies support that GBM has a poorer survival rate than LGG, the survival rates for patients with GBM and LGG are distinctly different. This characteristic creates a shortcoming in our data, as both GBM and LGG are unbalanced datasets. The classification accuracy may be affected by this imbalance. To achieve better accuracy, we applied the ROSE method to generate a more balanced dataset. The main idea of ROSE is to over-sample the minority group while sampling the minority group with replacement and sampling the majority without replacement. ROSE ensures that we have a fairly balanced data, allowing us to plot ROC curves and compare them to the unbalanced data.
Prediction model
After balancing the dataset, we used XGBoost and random forest to predict the survival status of glioma patients. XGBoost is a boosting method that builds multiple weak classifiers and enhances the model by weighting the parameters from weak classifiers. The direction of each weak classifier is to minimize the loss function using a gradient boosting algorithm. The main difference between XGBoost and random forest is that the trees in XGBoost are dependent on each other, whereas the trees in the random forest are independent. In random forest, trees are randomly selected from the samples and variables during the training process.
Statistical analysis
In this study, we conducted all data preprocessing and statistical analyses in R (4.2.2). The t-test was used to compare the difference in cuproptosis gene expression between GBM and LGG. The Kaplan–Meier curve was applied to compare the overall survival (OS) between GBM and LGG patients. When using machine learning methods such as random forest and XGBoost, data were randomly split into training and test sets in a 6:4 ratio. The unbalanced dataset was adjusted by applying the ROSE algorithm to achieve better prediction results.
Data availability
Data are available upon reasonable request from Peter Wang, Email: wangpeter2@hotmail.com.
Abbreviations
- GBM:
-
Glioblastoma
- GTEx:
-
Genotype-tissue expression
- GO:
-
Gene ontology
- LncRNA:
-
Long non-coding RNA
- LGG:
-
Low-grade glioma
- mRNA:
-
Messenger RNA
- ROSE:
-
Random over sampling examples
- TCGA:
-
The Cancer Genome Atlas Program
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- OS:
-
Overall survival
- PCA:
-
Principal component analysis
- WHO:
-
World Health Organization
References
Bale, T. A. & Rosenblum, M. K. The 2021 WHO Classification of Tumors of the Central Nervous System: An update on pediatric low-grade gliomas and glioneuronal tumors. Brain Pathol.32, e13060. https://doi.org/10.1111/bpa.13060 (2021).
Xu, S., Tang, L., Li, X., Fan, F. & Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett.476, 1–12. https://doi.org/10.1016/j.canlet.2020.02.002 (2020).
Ostrom, Q. T. et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro-oncology21, v1–v100. https://doi.org/10.1093/neuonc/noz150 (2019).
Berger, T. R., Wen, P. Y., Lang-Orsini, M. & Chukwueke, U. N. World Health Organization 2021 classification of central nervous system tumors and implications for therapy for adult-type gliomas: A review. JAMA Oncol.8, 1493–1501. https://doi.org/10.1001/jamaoncol.2022.2844 (2022).
Reifenberger, G., Wirsching, H.-G., Knobbe-Thomsen, C. B. & Weller, M. Advances in the molecular genetics of gliomas—implications for classification and therapy. Nat. Rev. Clin. Oncol.14, 434–452 (2017).
Velásquez, C. et al. Molecular and clinical insights into the invasive capacity of glioblastoma cells. J. Oncol.2019, 1740763. https://doi.org/10.1155/2019/1740763 (2019).
Nicholson, J. G. & Fine, H. A. Diffuse glioma heterogeneity and its therapeutic implications. Cancer Discov.11, 575–590. https://doi.org/10.1158/2159-8290.cd-20-1474 (2021).
Richard, Q., Laurenge, A., Mallat, M., Sanson, M. & Castro-Vega, L. J. New insights into the immune TME of adult-type diffuse gliomas. Curr. Opin. Neurol.35, 794–802. https://doi.org/10.1097/wco.0000000000001112 (2022).
Kumthekar, P., Raizer, J. & Singh, S. Low-grade glioma. Current Understanding and Treatment of Gliomas, 75–87 (2015).
Yang, K. et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer21, 39. https://doi.org/10.1186/s12943-022-01513-z (2022).
Jia, K. W. et al. Interferon-alpha stimulates DExH-box helicase 58 to prevent hepatocyte ferroptosis. Mil Med. Res.11, 22. https://doi.org/10.1186/s40779-024-00524-9 (2024).
Liu, D., Qin, H., Gao, Y., Sun, M. & Wang, M. Cardiovascular disease: Mitochondrial dynamics and mitophagy crosstalk mechanisms with novel programmed cell death and macrophage polarisation. Pharmacol. Res.206, 107258. https://doi.org/10.1016/j.phrs.2024.107258 (2024).
Zhou, S., Liu, J., Wan, A., Zhang, Y. & Qi, X. Epigenetic regulation of diverse cell death modalities in cancer: A focus on pyroptosis, ferroptosis, cuproptosis, and disulfidptosis. J. Hematol. Oncol.17, 22. https://doi.org/10.1186/s13045-024-01545-6 (2024).
Su, P. P. et al. Down-regulation of Risa improves podocyte injury by enhancing autophagy in diabetic nephropathy. Mil Med. Res.9, 23. https://doi.org/10.1186/s40779-022-00385-0 (2022).
Cobine, P. A. & Brady, D. C. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol. Cell82, 1786–1787. https://doi.org/10.1016/j.molcel.2022.05.001 (2022).
Tang, D., Chen, X. & Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res.32, 417–418. https://doi.org/10.1038/s41422-022-00653-7 (2022).
Kahlson, M. A. & Dixon, S. J. Copper-induced cell death. Science375, 1231–1232. https://doi.org/10.1126/science.abo3959 (2022).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science375, 1254–1261 (2022).
Wang, Y., Zhang, L. & Zhou, F. Cuproptosis: A new form of programmed cell death. Cellular & Molecular Immunology, 1–2 (2022).
Wick, W., Osswald, M., Wick, A. & Winkler, F. Treatment of glioblastoma in adults. Ther. Adv. Neurol. Disord.11, 1756286418790452. https://doi.org/10.1177/1756286418790452 (2018).
Mudassar, F., Shen, H., O’Neill, G. & Hau, E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J. Exp. Clin. Cancer Res.39, 208. https://doi.org/10.1186/s13046-020-01724-6 (2020).
Xin, S. et al. Identification and quantification of necroptosis landscape on therapy and prognosis in kidney renal clear cell carcinoma. Front. Genet.13, 832046 (2022).
Yan, X. et al. A cuproptosis-related lncRNAs signature for prognosis, chemotherapy, and immune checkpoint blockade therapy of low-grade glioma. Front. Mol. Biosci.9, 966843 (2022).
Shen, L. P. et al. Advances in the mechanism of small nucleolar RNA and its role in DNA damage response. Mil Med. Res.11, 53. https://doi.org/10.1186/s40779-024-00553-4 (2024).
Hussen, B. M. et al. Targeting miRNA by CRISPR/Cas in cancer: Advantages and challenges. Mil. Med. Res.10, 32. https://doi.org/10.1186/s40779-023-00468-6 (2023).
Yang, Z., Gong, W., Zhang, T. & Gao, H. Molecular features of glioma determined and validated using combined TCGA and GTEx data analyses. Front. Oncol.11, 729137 (2021).
Wang, L. et al. Identification of cuproptosis-related lncRNAs for prognosis and immunotherapy in glioma. J. Cell. Mol. Med.26, 5820–5831 (2022).
Miao, Y. et al. Machine learning identification of cuproptosis and necroptosis-associated molecular subtypes to aid in prognosis assessment and immunotherapy response prediction in low-grade glioma. Front. Genet.13, 951239 (2022).
Jiang, R. et al. Transcriptional and genetic alterations of cuproptosis-related genes correlated to malignancy and immune-infiltrate of esophageal carcinoma. Cell Death Discov.8, 1–13 (2022).
Ouyang, Z., Zhang, H., Lin, W., Su, J. & Wang, X. Bioinformatic profiling identifies the glutaminase to be a potential novel cuproptosis-related biomarker for glioma. Front. Cell Dev. Biol.10, 982439. https://doi.org/10.3389/fcell.2022.982439 (2022).
Tang, S. et al. Identification of a novel cuproptosis-related gene signature for prognostic implication in head and neck squamous carcinomas. Cancers14, 3986 (2022).
Grier, J. T. & Batchelor, T. Low-grade gliomas in adults. Oncologist11, 681–693 (2006).
Li, G. et al. A novel DNA repair-related nomogram predicts survival in low-grade gliomas. CNS Neurosci. Ther.27, 186–195. https://doi.org/10.1111/cns.13464 (2021).
Finch, A. et al. Advances in research of adult gliomas. Int. J. Mol. Sci.22, 924. https://doi.org/10.3390/ijms22020924 (2021).
Lang, F., Liu, Y., Chou, F. J. & Yang, C. Genotoxic therapy and resistance mechanism in gliomas. Pharmacol. Ther.228, 107922. https://doi.org/10.1016/j.pharmthera.2021.107922 (2021).
Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol.131, 803–820. https://doi.org/10.1007/s00401-016-1545-1 (2016).
Zeiner, P. S. et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol.29, 513–529. https://doi.org/10.1111/bpa.12690 (2019).
Ghiaseddin, A. P., Shin, D., Melnick, K. & Tran, D. D. Tumor treating fields in the management of patients with malignant gliomas. Curr. Treat. Opt. Oncol.21, 76. https://doi.org/10.1007/s11864-020-00773-5 (2020).
Ghouzlani, A. et al. Immune checkpoint inhibitors in human glioma microenvironment. Front. Immunol.12, 679425. https://doi.org/10.3389/fimmu.2021.679425 (2021).
Chen, S. et al. Prognostic value of cuproptosis-related genes signature and its impact on the reshaped immune microenvironment of glioma. Front. Pharmacol.13, 1016520. https://doi.org/10.3389/fphar.2022.1016520 (2022).
Cortes, J. et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. New Engl. J. Med.387, 217–226. https://doi.org/10.1056/NEJMoa2202809 (2022).
Zhang, C., Wang, L., Guo, Y. & Feng, W. Systematic analysis of brain and skull ischemic injury expression profiles reveals associations of the tumor immune microenvironment and cell death with ischemic stroke. Front. Immunol.13, 1082546. https://doi.org/10.3389/fimmu.2022.1082546 (2022).
Chen, B. et al. A Cuproptosis Activation Scoring model predicts neoplasm-immunity interactions and personalized treatments in glioma. Comput. Biol. Med.148, 105924. https://doi.org/10.1016/j.compbiomed.2022.105924 (2022).
Bao, J. H. et al. Identification of a novel cuproptosis-related gene signature and integrative analyses in patients with lower-grade gliomas. Front. Immunol.13, 933973. https://doi.org/10.3389/fimmu.2022.933973 (2022).
Ye, Z. et al. Development and validation of cuproptosis-associated prognostic signatures in WHO 2/3 glioma. Front. Oncol.12, 967159. https://doi.org/10.3389/fonc.2022.967159 (2022).
Zhang, Z., Wang, B., Xu, X. & Xin, T. Cuproptosis-related gene signature stratifies lower-grade glioma patients and predicts immune characteristics. Front. Genet.13, 1036460. https://doi.org/10.3389/fgene.2022.1036460 (2022).
Wang, L. et al. Identification of cuproptosis-related lncRNAs for prognosis and immunotherapy in glioma. J. Cell Mol. Med.26, 5820–5831. https://doi.org/10.1111/jcmm.17603 (2022).
Chen, D. et al. A novel signature of cuproptosis-related lncRNAs predicts prognosis in glioma: Evidence from bioinformatic analysis and experiments. Front. Pharmacol.14, 1158723. https://doi.org/10.3389/fphar.2023.1158723 (2023).
Jin, J. et al. Studies on the therapeutic and prognostic biomarkers of glioma using a novel cuproptosis-related IncRNA signature and validation in glioma. J. Environ. Pathol. Toxicol. Oncol.42, 53–70. https://doi.org/10.1615/JEnvironPatholToxicolOncol.2023047159 (2023).
Wen, J., Zhao, W. & Shu, X. A novel cuproptosis-related LncRNA signature: Prognostic and therapeutic value for low grade glioma. Front. Oncol.12, 1087762. https://doi.org/10.3389/fonc.2022.1087762 (2022).
Yan, X. et al. A cuproptosis-related lncRNAs signature for prognosis, chemotherapy, and immune checkpoint blockade therapy of low-grade glioma. Front. Mol. Biosci.9, 966843. https://doi.org/10.3389/fmolb.2022.966843 (2022).
Xu, Y., Wang, C., Li, S., Zhou, H. & Feng, Y. Prognosis and immune response of a cuproptosis-related lncRNA signature in low grade glioma. Front. Genet.13, 975419. https://doi.org/10.3389/fgene.2022.975419 (2022).
Zhang, B., Chen, J., Cui, M. & Jiang, Y. LncRNA ZFAS1/miR-1271-5p/HK2 promotes glioma development through regulating proliferation, migration, invasion and apoptosis. Neurochem. Res.45, 2828–2839. https://doi.org/10.1007/s11064-020-03131-x (2020).
Li, X. et al. The long noncoding RNA ZFAS1 promotes the progression of glioma by regulating the miR-150-5p/PLP2 axis. J. Cell Physiol.235, 2937–2946. https://doi.org/10.1002/jcp.29199 (2020).
Gao, K., Ji, Z., She, K., Yang, Q. & Shao, L. Long non-coding RNA ZFAS1 is an unfavourable prognostic factor and promotes glioma cell progression by activation of the Notch signaling pathway. Biomed. Pharmacother.87, 555–560. https://doi.org/10.1016/j.biopha.2017.01.014 (2017).
Lv, Q. L. et al. Upregulation of long noncoding RNA zinc finger antisense 1 enhances epithelial-mesenchymal transition in vitro and predicts poor prognosis in glioma. Tumour Biol.39, 1010428317695022. https://doi.org/10.1177/1010428317695022 (2017).
Anjom-Shoae, J. et al. Dietary insulin index and insulin load in relation to glioma: Findings from a case–control study. Nutr. Neurosci.24, 354–362 (2021).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci.28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res.51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Funding
This project was supported by the Ningxia Natural Science Foundation Project (2022AAC03519), Key R&D Project of Ningxia Hui Autonomous Region (2020BEG3017), Beijing Zhongwei Biological Research Transformation Research Center Project (ZWYX20230002) and Project of Dongguan First Hospital Affiliated to Guangdong Medical University (GCC202402).
Author information
Authors and Affiliations
Contributions
GH and LS designed this study. SH, MG, QL performed the analysis. SH and QL wrote the manuscript. GH, LS and PW edited the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, S., Gao, M., Li, Q. et al. Machine learning predicts cuproptosis-related lncRNAs and survival in glioma patients. Sci Rep 14, 22323 (2024). https://doi.org/10.1038/s41598-024-72664-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-72664-w
Keywords
This article is cited by
-
A novel approach to enhance glioblastoma multiforme treatment efficacy: non-coding RNA targeted therapy and adjuvant approaches
Clinical Epigenetics (2025)
-
Multidimensional analysis reveals gene expression, cell interactions, and signaling networks in glioma and Alzheimer’s disease
Discover Oncology (2025)
