
Abstract
The epidemiology and circulation patterns of various rhinovirus types within populations remains under-explored. We generated 803 VP4/VP2 gene sequences from rhinovirus-positive samples collected from acute respiratory illness (ARI) patients, including both in-patient and outpatient cases, between 1st January and 31st December 2014 from eleven surveillance sites across Kenya and used phylogenetics to characterise virus introductions and spread. RVs were detected throughout the year, with the highest detection rates observed from January to March and June to July. We detected a total of 114 of the 169 currently classified types. Our analysis revealed numerous virus introductions into Kenya characterized by local expansion and extinction, and extensive spatial mixing of types within the country due to the widespread transmission of the virus after an introduction. This work demonstrates that in a single year, the circulation of rhinovirus in Kenya was characterized by substantial genetic diversity, multiple introductions, and extensive geographical spread.
Similar content being viewed by others

Introduction
Rhinoviruses (RVs) are one of the leading cause of acute upper respiratory tract illnesses, commonly referred to as “common cold”1,2. RV infections occur all year, although they peak in early autumn and late spring in many temperate countries, and in the rainy seasons in tropical countries2,3. RVs cause 8–12 episodes of respiratory infections in children and 2–4 episodes in adults each year4,5,6, posing significant social and economic burden due to time lost from work or school, medical attendance, and reduced performance of regular duties7. Despite the high prevalence and clinical importance of RV infections, little is known about the patterns of occurrence of RV types, as well as the role of virus type in the RV distribution.
RVs belong to the genus Enterovirus of the family Picornaviridae. The virus genome consists of ~ 7200 nucleotides and encodes four structural proteins (VP4, VP2, VP3 and VP1) and seven non-structural proteins (2 A, 2B, 2 C, 3 A, 3B, 3 C and 3D)2. The single-stranded positive sense RNA genome is enclosed within a protein capsid that is made up of sixty copies of each of the four structural proteins, of which VP1, VP2, and VP3 are exposed outside, while VP4 is completely masked in the capsid2,8,9. The three surface-exposed capsid proteins carry the antigenically important sites2,8,9. Genetic variation in the VP4/VP2 and VP1 genomic regions has been useful in molecular typing10,11 and epidemiological investigations12,13,14. As of 15th March 2023, a total of 169 RV types have been described and classified into three distinct species, i.e., Rhinovirus A, Rhinovirus B, and Rhinovirus C15.
Viral sequence data are increasingly used to track the geographic spread and transmission of viral pathogens16,17. Genomic data has considerably informed public health interventions and outbreak management for viral pathogens, e.g., SARS-CoV-2, Ebolavirus, and Zika virus18,19,20,21. Widespread spatial and temporal transmission patterns of RV were previously described within the Kilifi Health and Demographic Surveillance System (KHDSS) on the Kenyan Coast covering around 300,000 residents22 and marked by multiple virus introductions12. Transmission patterns of RV at a nationwide scale have not been documented in Kenya. Here we analyze 803 sequences obtained from individuals presenting with acute respiratory illness (ARIs) at 11 sentinel surveillance sites across Kenya in 2014 23, to explore the temporal and spatial circulation patterns of RV types at the countrywide scale.
Methods
Ethical considerations
The study was approved by the Kenya Medical Research Institute - Scientific and Ethical Review Unit (SSC #3044) and CDC Institutional Review Board (#6806) to use pre-existent, pseudonymized specimens, and data. All individuals, parents and guardians gave written informed consent for themselves or their children to participate in the original studies23. For older children aged 13–17 years, assent was obtained as part of the individual informed consenting process. All the experiments in this study were performed in accordance with relevant guidelines and regulations.
Surveillance sites
This study utilised samples collected throughout the surveillance period covering 1st January to 31st December 2014 from three health facility-based respiratory virus surveillance programs in Kenya (Table 1; Fig. 1). The first program included eight inpatient hospitals participating in the influenza sentinel surveillance: Dadaab Refugee Camp (RC), Kakuma RC, Kenyatta National Hospital (KNH), Nakuru County Referral Hospital (CRH), Kakamega CRH, Nyeri CRH, Siaya CRH and Coast General Teaching and Referral Hospital24,25,26; the second included two outpatient clinics; Lwak Mission Hospital, Asembo and Tabitha Medical Clinic, Kibera under Population-Based Infectious Disease Surveillance (PBIDS)27, and the third program was a pediatric inpatient sampling at the Kilifi County Hospital (KCH)28. The influenza sentinel surveillance was established by Kenya Medical Research Institute-Centre for Global Research (KEMRI-CGHR), the US Centers for Disease Control and Prevention (CDC)-Kenya, and the Ministry of Health as part of the Global Influenza Surveillance and Response System since 200624. The PBIDS platform in Asembo and Kibera is run by KEMRI-CGHR with financial and technical support from CDC since 200629. Surveillance at KCH is conducted by the KEMRI-Centre for Geographic Medical Research Coast (CGMRC) under KEMRI-Wellcome Trust Research Programme (KWTRP) in Kilifi, Kenya28. Overall, the selected surveillance sites offer a good representation of the various geographical settings in Kenya.

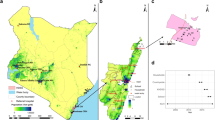
Map of Kenya showing the geo-location of the 11 respiratory surveillance sites, distinguishing inpatient (red markers) from outpatient (green markers) facilities.
Patient enrollment
The surveillance sites recruited patients of various ages presenting with acute respiratory illness (ARI) with a measured fever of ≥ 38 °C and a cough with an onset of symptoms within the last 7 days (influenza-like illness, ILI) or acute respiratory illness requiring hospitalization with a history of fever or measured fever ≥ 38 °C and a cough with an onset of symptoms within the last 14 days severe acute respiratory illness, (SARI), or acute lower respiratory tract illness (ALRTI) defined as the presence of cough OR difficulty in breathing with one of the following danger signs: chest in-drawing, stridor, unable to breastfeed, vomit everything, convulsions, lethargy, or unconsciousness or an adaptation of WHO severe/very severe pneumonia as described in a Table 1. The different case definitions were consistently applied within each platform over the course of the study period.
Data and specimen collection
Patient demographic data and clinical features of presenting illness were collected in real-time using custom designed databases. Nasopharyngeal (NP) and/or oropharyngeal (OP) swabs were collected from eligible patients who presented to health facilities with clinical features of an acute respiratory illness as describe in patient enrolment section. NP/OP samples were stored in viral transport media, and transported to the laboratory for long-term storage at -80oC.
RV screening and sequencing
Viral RNA was extracted using RNeasy Mini Kit (Qiagen, Germany) in Qiacube HT as per the manufacturer’s instructions and screened for respiratory viruses (i.e. RSV (groups A and B), rhinovirus, human coronaviruses (hCoV-OC43, -NL63, -229E), influenza A virus and adenovirus) using a multiplex real-time reverse-transcription PCR (rt-RT-PCR) assay30,31. A sample was considered RV positive if the rt-RT-PCR cycle threshold (Ct) was ≤ 35.032. The VP4/VP2 viral genomic region (~ 420 nucleotides long) of positive samples was amplified and sequenced as previously described12. Consensus sequences were assembled using the Sequencher software version 5.4.6 (Gene Codes Corporation, Ann Arbor, USA).
RV species and type assignment
We used the term ‘type’ to refer to RV sequences classified as distinct by genetic comparisons as described previously11. Sequences were assigned into the same RV type based on > 90% nucleotide similarity to rhinovirus prototype sequences (also referred to as reference sequences11 and phylogenetic clustering with bootstrap support value > 70%11. Distributions of pairwise genetic distances were assessed for evaluation of intertype and intra-type divergence11. Intra-type ‘variant’ was defined based on a divergence cut-off or threshold value determined as the least frequent value between the first (same phylogenetic clade) and second (different phylogenetic clades) modes in a pairwise nucleotide difference distribution plot as previously described33. Viruses within the same phylogenetic clade were assumed to belong to the same variant of an RV type.
Global dataset
Rhinovirus VP4/VP2 sequences from other regions around the world deposited in GenBank as of 31 December 2021, whose sequenced regions overlapped the 803 Kenyan sequences generated in this study and derived from viruses sampled between 1st January 2005 and 31st December 2014, were collated and phylogenetically compared with the Kenya virus sequences. The complete comparison global data set comprised 4,448 VP4/VP2 sequences from 40 countries, including 653 sequences from Kenya (Kilifi (n = 627), Nairobi (n = 10), Mombasa (n = 4), Malindi (n = 3), Alupe (n = 3), Kisumu (n = 2), Isiolo (n = 2), Kisii (n = 1) and Kericho (n = 1) (Appendix Table 1).
Phylogenetic analysis
Multiple sequence alignments (MSA) were generated using MAFFT v7.22034 and maximum likelihood phylogenetic trees estimated using IQ-TREE v1.6.1235. Branch support was assessed by 1000 bootstrap iterations. The temporal signal in the data was examined using TempEst v1.5.336. TreeTime v.0.8.1 was used to transform the ML tree topologies into dated trees. Phylogenetic trees were visualized using ggtree v2.2.4 package37 in R38.
Statistical analysis
Statistical analysis was conducted using STATA version 15.1 (College Station, Texas). Chi-square test was used to assess the association between the different ARI definitions, care settings and age categories with the HRV species. Site-specific monthly percent virus positivity was computed.
Type diversity calculation
RV type diversity was calculated using diversity indices (Shannon (H) and Simpson (D))39,40. The calculations were as follows:
Where p is the proportion (n/N) of individual types found in one individual species (n) divided by the total number of types found (N), and s is the total number of species. Higher scores of H and D indicates high diversity.
Results
Between 1st January and 31st December 2014, a total of 6398 NP and/or OP swabs were collected from patients who voluntarily presented to the eleven facilities with ARI. Testing was done on 5859 (91.6%) available samples, Table 2. Of the tested samples, 5665 (96.7%) were linked with their respective demographic and clinical data. Two hundred and fourteen samples (214) collected from Siaya and Kibera patients with missing data on respiratory symptoms were excluded from the study, the final analytical dataset comprised of 5451 specimens. RV was detected in 17.0% (924/5451) of samples collected from the 11 surveillance sites (Table 2). Of the 924 positive samples, 147 (15.9%) were co-infected with multiple viruses, such as RSV A/B, adenoviruses, hCoVs, and influenza A viruses. The percent of samples that were virus positive for RV infections varied by surveillance site; Siaya recorded the highest positives (24.0%) while Dadaab (10.4%) recorded the lowest (Table 2). Rhinovirus was detected throughout the year in most sites, although the prevalence of detections varied by month of sampling due to seasonal variation in SARI (Fig. 2). Different sites experienced peak occurrence in different months, but a majority saw infection peaks between January - March (7/11) and June - July (5/11) (Fig. 2). There was no RV detected in April in Kenyatta National Hospital (KNH) and in May in Kakuma and Kibera sites (Fig. 2). There were no samples collected in Dadaab between July and September.

Monthly distribution of samples tested and cases positives for rhinovirus from the 11 surveillance sites in Kenya in 2014.
VP4/VP2 sequences were successfully obtained from 803/924 (87.0%) RV positive samples (Table 2). The remaining samples either failed to amplify with the VP4/VP2 specific primers (n = 111) or were identified as non-RV enteroviruses (n = 10). The RT-PCR cycle threshold values for the samples that failed sequencing ranged from 23.98 to 35.00. Overall, 492 (61.3%) sequences were classified as Rhinovirus A comprising 58 types; 63 (7.8%) sequences were Rhinovirus B comprising 16 types, and 248 (30.9%) were Rhinovirus C comprising 40 types. The most detected types were A34 (n = 35), A22 (n = 30), A58 (n = 27), A12 (n = 25) and A78 (n = 23) (Supplementary Table 1). The number of types largely reflected the number of samples sequenced and varied between sites (Supplementary Fig. 1A). Comparison by age categories (paediatric vs. adults) or ARI definition (i.e., ILI vs. SARI vs. ALRTI) across different RV types revealed significant differences in age categories (p-value < 0.001) and ARI definition (p-value = 0.03) (Table 3). Rhinovirus type diversity per site calculated using the Shannon (H) and Simpson (D) indices indicated that Siaya had the highest value of diversity, followed by Lwak and Kilifi. Dadaab reported the lowest value of diversity (Table 4). All three RV species were detected in all sites, except for Dadaab, where Rhinovirus B was not detected (Supplementary Fig. 1B). The proportions of RV species were similar in all the sites; Rhinovirus A (range, 51–75%) and Rhinovirus C (range, 17–46%) were frequently detected, while Rhinovirus B (range, 0–13%) infections were low or not detected (Supplementary Fig. 1B).
Spatial-temporal distribution of RV types in Kenya
We identified up to 49 unique RV types co-circulating in a single month across the country, and/or up to 20 unique types within a single location in a single month (Supplementary Fig. 2). The duration of circulation varied by RV type; several types occurred at a high prevalence and for consecutive months, while others occurred once or intermittently during the study period (Supplementary Fig. 3). For example, RV-A22 was detected throughout the year, A34 was present for 11 consecutive months (February to December 2014), and RV-C7 and A21 circulated consecutively for 8 months (February to September and May to December, respectively) (Supplementary Fig. 3). Spatially, several RV types circulated widely; some circulated in multiple sites during the same timeframe, while others circulated in multiple sites at different times (Fig. 3 and Supplementary Fig. 4). For example, RV-A34 was detected in all 10 sites (Fig. 3), while A22 was detected in 11 sites (Supplementary Fig. 4). We observed the occurrence of localized type-specific epidemics or outbreaks; the distribution of RV types was similar between neighbouring locations (Supplementary Fig. 4). For example, types A21 and A78 circulated in Asembo, Siaya, and Kakamega in the same period (June-July), while A49 was seen in Mombasa and Kilifi between July and December (Supplementary Fig. 4). Other types were more random in occurrence with no discernable temporal or spatial pattern.

Temporal distributions of frequent RV A34 across different sites.
Phylogenetic clustering
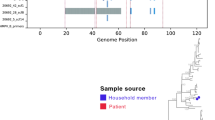
We reconstructed time-scaled phylogenies for thirteen prevalent types. In the global context, Kenyan viruses formed monophyletic clusters/clades containing sequences from different sampling sites in Kenya, suggesting multiple introductions and local transmission chains (Fig. 4 and Supplementary Fig. 5). For example, A34 viruses collected in 2014 separated into 2 major clusters, each with sequences from different sites (Fig. 4). Similarly, the Kenyan RV-A22 viruses collected between 2008 and 2014 separated into 3 major clusters, each comprising sequences from different locations in Kenya (Supplementary Fig. 5). Similar observations were observed for other RV types including, A12, A49, A58, A78, and C10 (Supplementary Fig. 5). Certain clades or clusters were location-specific and genetically distinct from other Kenyan 2014 sequences. For example, an A78 variant from Nyeri and an A58 variant in Dadaab (Supplementary Fig. 5), showing evidence of localised transmission clusters.

Time-resolved lineage-specific phylogenetic trees for RV A34. The Kenya genomes are indicated with filled circles coloured by site.
Sequences originating from Kenya did not form a single monophyletic group on the phylogenetic tree, instead, they were interspersed mostly as singleton or clusters located separately or with clusters of viruses from other countries. For example, RV-A22 sequences from Kenya clustered closely with sequences from China and France in 2014, while A12 sequences clustered closely with sequences from China, the USA, France, Uganda, and Nepal in 2014. The Kenyan A58 sequences clustered closely with those from USA and France, while the A78 sequences clustered closely with Chinese and American sequences, and the A34 resembled Nepalese sequences.
Discussion
This study provides a detailed description of the spatial-temporal dynamics of introductions and spread of rhinovirus in Kenya based on phylogenetic analyses. We show that the persistent circulation of RV in Kenya was mostly driven by multiple introductions of different types throughout the one-year period, leading to established local transmission.
Rhinovirus A was the predominant species circulating in Kenya in 2014, followed by Rhinovirus C, and the least common was Rhinovirus B. This is consistent with a previous surveillance study across 8 sampling sites in Kenya in 200841. Although differences in subject recruitment strategies may not allow one-on-one comparison between this study and others, these observations are similar to studies reported in Africa, USA, Asia, Europe, the Middle East, and Australia11,42. The similarities in frequencies of RV species in diverse geographies would suggest that RV circulates unrestricted globally.
RV was detected all year, with the highest detection rates observed in January to March and June to July. The year-round occurrence of rhinovirus in the country was largely sustained by contemporaneous and successive mini epidemics or outbreaks caused by distinct types and variants that were probably introduced separately into the country or diversified locally after a single introduction.
Spatial-temporal analysis revealed occurrence of multiple types and variants across space and time in agreement with previous studies12,14,43. The observed variations in time and duration of circulation among the rhinovirus types could be due to (i) differences in the duration of type-specific immunity, (ii) frequency of introductions or, iii) level of antigenic similarity (heterologous immunity)33. The concurrent and sequential circulation of RV types seen as the occurrence of multiple peaks of the same type in the same or distant locations could signify multiple introductions, antigenic variation, or infections in different population strata44. The heterogeneity in RV detection by month and location could be attributed to the seasonal variation in SARI or sampling methods or to regional differences in environmental and climatic factors. Climatic factors such as temperature, rainfall, and relative humidity have been hypothesized to influence RV activity in the tropics3,45,46.
Even with non-uniform sampling and short duration of study (1 year), we showed close genetic association among sequences from different sites as a result of widespread transmission of the virus in the country. On a finer spatial scale, study sites occurring within the same geographical region (as described in Table 1) had a similar distribution of RV types, an indicator of a point source outbreak, in which a single virus type enters a location and diffuses through the interconnected populations probably sharing social amenities12. This agrees with our previous work that showed greater similarity in RV types among the locations in proximity12.
The global phylogenies showed that RV viruses circulating in Kenya were closely related to strains circulating in Europe, Asia, and North America. The Kenyan RV diversity appears to be nested within the global diversity as a result of transmission facilitated by unrestricted movement, increased connectivity, and social mixing.
Although our analyses were limited to the VP4/VP2 genomic region, we highlight the use of sequence data to trace the introduction and spread of rhinovirus at the countrywide level and show the benefit of systematic, continuous, and geographically representative surveillance to detect and monitor the occurrence of types at a larger scale. Whole genome sequencing could provide more insight into virus diversity and transmission47, and we recommend that future studies should combine genomic data with epidemiological and anthropological data (e.g., host migration, immunity profiles, population densities, and social contact patterns) to further elucidate patterns of RV infections.
This study had some limitations. First, due to the retrospective approach of the study, it was not possible to recover sequences from all the samples due to sample degradation; samples (n = 111/924) that failed sequencing had considerably higher Ct values (low viral load) compared to samples that were successfully sequenced with low Ct values (high viral load). Second, there were varying study design by sites, for instance, the disease case definition and ages enrolled. The varying designs would mainly affect the virus positivity estimates observed over the one-year surveillance period, in that sites that had more inclusive case definition that includes mild cases of respiratory illnesses may exhibit higher rates of rhinovirus positivity compared to sites using a more restrictive case definition that only includes severe cases of respiratory illnesses. We also acknowledge the potential issue with using within-14 day specimens collected from SARI patients, especially concerning the differential shedding periods of various viruses. Unfortunately, we do not have data on the exact number of days between symptom onset and specimen collection. Third, the inclusion of fever as a criterion in most of the case definitions used may lead to the underestimation of RV prevalence since many RV infections, including severe cases, do not necessarily manifest with fever. Consequently, there is a possibility that we might have missed RV patients who do not exhibit fever symptoms, potentially impacting the overall assessment of RV prevalence in our study. Fourth, the short sequence fragment analysed in this study may result in spurious phylogenetic connections. Lastly, the study focused on samples that were collected in hospital settings, which may have missed genotypes associated with asymptomatic or mildly ill infections.
In conclusion, our study demonstrates that sustained circulation of the virus in 2014 was due to frequent introductions of different types and variants into the country followed by local spread for some of these introductions. Temporal differences in persistence of rhinovirus types over the one-year period, could be attributed to differences in the frequency and number of virus introductions into the country. Spatial patterns show extensive spread of the virus, and the evidence of similar distribution of types in locations that are in proximity to each other may indicate local partitioning or spatial structures of virus transmission.
Data availability
Sequence data generated in this study are available in GenBank under accession numbers: MZ129390 - MZ130096. Additional data and analysis scripts for this manuscript are available at the VEC Harvard Dataverse: https://doi.org/10.7910/DVN/CBHVTA.
References
Ouédraogo, S. et al. Viral etiology of respiratory tract infections in children at the Pediatric Hospital in Ouagadougou (Burkina Faso). PLoS One. 9 (10), e110435 (2014).
Jacobs, S. E., Lamson, D. M., St George, K. & Walsh, T. J. Human rhinoviruses. Clin. Microbiol. Rev.26 (1), 135–162. https://doi.org/10.1128/CMR.00077-12 (2013).
Garcia, J. et al. Human rhinoviruses and enteroviruses in influenza-like illness in Latin America. Virol. J.10, 305. https://doi.org/10.1186/1743-422X-10-305 (2013).
Nguyen, D. N. T. et al. Epidemiology and etiology of influenza-like-illness in households in Vietnam; it’s not all about the kids! J. Clin. Virol.82, 126–132. https://doi.org/10.1016/j.jcv.2016.07.014 (2016).
Kamau, E. et al. An intensive, active surveillance reveals continuous Invasion and High Diversity of Rhinovirus in households. J. Infect. Dis.219 (7), 1049–1057. https://doi.org/10.1093/infdis/jiy621 (2019).
Arruda, E., Pitkaranta, A., Witek, T. J. J., Doyle, C. A. & Hayden, F. G. Frequency and natural history of rhinovirus infections in adults during autumn. J. Clin. Microbiol.35 (11), 2864–2868 (1997).
Fendrick, A. M., Monto, A. S., Nightengale, B. & Sarnes, M. The economic burden of non-influenza-related viral respiratory tract infection in the United States. Arch. Intern. Med.163 (4), 487–494 (2003). doi:ioi20041 [pii].
Esneau, C., Bartlett, N. & Bochkov, Y. A. Chapter 1 - Rhinovirus structure, replication, and classification. In: Bartlett N, Wark P, Knight D, eds. Rhinovirus Infections. Academic Press; :1–23. doi: (2019). https://doi.org/10.1016/B978-0-12-816417-4.00001-9
Stobart, C. C., Nosek, J. M. & Moore, M. L. Rhinovirus Biology, antigenic diversity, and advancements in the design of a human rhinovirus vaccine. Front. Microbiol.8https://doi.org/10.3389/fmicb.2017.02412 (2017).
Simmonds, P. et al. Proposals for the classification of human rhinovirus species C in genotypically assigned types. J. Gen. Virol.91https://doi.org/10.1099/vir.0.023994-0 (2010). LB - Simmonds2010.
McIntyre, C. L., Knowles, N. J. & Simmonds, P. Proposals for the classification of human rhinovirus species a, B and C into genotypically assigned types. J. Gen. Virol.94 (Pt 8), 1791–1806. https://doi.org/10.1099/vir.0.053686-0 (2013).
Morobe, J. M. et al. Human rhinovirus spatial-temporal epidemiology in rural coastal Kenya, 2015–2016, observed through outpatient surveillance [version 2; peer review: 2 approved]. Wellcome Open. Res.3 (128). https://doi.org/10.12688/wellcomeopenres.14836.2 (2019).
Arakawa, M. et al. Molecular epidemiological study of human rhinovirus species a, B and C from patients with acute respiratory illnesses in Japan. J. Med. Microbiol.61, 410–419. https://doi.org/10.1099/jmm.0.035006-0 (2012).
van der Linden, L. et al. A molecular epidemiological perspective of rhinovirus types circulating in Amsterdam from 2007 to 2012. Clin. Microbiol. Infect.22 (12), 1002. .e9-1002.e14 (2016).
The Picornavirus Pages. Enteroviruses. Enteroviruses. Accessed March 15, 2023. (2006). https://www.picornaviridae.com/ensavirinae/enterovirus/enterovirus.htm
Grubaugh, N. D. et al. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol.4 (1), 10–19. https://doi.org/10.1038/s41564-018-0296-2 (2019).
Gardy, J. L. & Loman, N. J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet.19 (1), 9–20. https://doi.org/10.1038/nrg.2017.88 (2018).
Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228–232. https://doi.org/10.1038/nature16996 (2016).
Eduan, W. et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Sci. (1979). 374 (6566), 423–431. https://doi.org/10.1126/science.abj4336 (2021).
Gire, S. K. et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014;345:1369–1372. doi: (1979). https://doi.org/10.1126/science.1259657
Faria, N. R. et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature. 546 (7658), 406–410. https://doi.org/10.1038/nature22401 (2017).
Scott, J. A. G. et al. Profile: the Kilifi Health and demographic Surveillance System (KHDSS). Int. J. Epidemiol.41 (3), 650–657. https://doi.org/10.1093/ije/dys062 (2012).
Murunga, N. et al. Surveillance of respiratory viruses at health facilities from across Kenya, 2014 [version 2; peer review: 1 approved with reservations]. Wellcome Open. Res.7 (234). https://doi.org/10.12688/wellcomeopenres.17908.2 (2022).
Katz, M. A. et al. Results from the First Six years of National Sentinel Surveillance for Influenza in Kenya, July 2007–June 2013. PLoS One. 9 (6), e98615. https://doi.org/10.1371/journal.pone.0098615 (2014).
Bigogo, G. M. et al. Epidemiology of respiratory syncytial virus infection in Rural and Urban Kenya. J. Infect. Dis.208 (suppl_3), S207–S216. https://doi.org/10.1093/infdis/jit489 (2013).
Emukule, G. O. et al. The Burden of Influenza and RSV among inpatients and outpatients in Rural Western Kenya, 2009–2012. PLoS One. 9 (8), e105543. https://doi.org/10.1371/journal.pone.0105543 (2014).
Feikin, D. R. et al. The Burden of Common Infectious Disease syndromes at the Clinic and Household Level from Population-based Surveillance in Rural and Urban Kenya. PLoS One. 6 (1), e16085. https://doi.org/10.1371/journal.pone.0016085 (2011).
Nokes, D. J. et al. Incidence and severity of respiratory syncytial virus pneumonia in rural Kenyan children identified through hospital surveillance. Clin. Infect. Dis.49 (9), 1341–1349. https://doi.org/10.1086/606055 (2009).
Feikin, D. R. et al. Etiology and incidence of viral and bacterial Acute Respiratory illness among older children and adults in Rural Western Kenya, 2007–2010. PLoS One. 7 (8), e43656. https://doi.org/10.1371/journal.pone.0043656 (2012).
Gunson, R. N., Collins, T. C. & Carman, W. F. Real-time RT-PCR detection of 12 respiratory viral infections in four triplex reactions. J. Clin. Virol.33 (4), 341–344. https://doi.org/10.1016/j.jcv.2004.11.025 (2005).
Hammitt, L. L. et al. Added value of an oropharyngeal swab in detection of viruses in children hospitalized with lower respiratory tract infection. J. Clin. Microbiol.49 (6), 2318–2320. https://doi.org/10.1128/JCM.02605-10 (2011).
Onyango, C. O. et al. Molecular epidemiology of human rhinovirus infections in Kilifi, coastal Kenya. J. Med. Virol.84 (5), 823–831. https://doi.org/10.1002/jmv.23251 (2012).
Mwita Morobe, J. et al. Trends and Intensity of Rhinovirus invasions in Kilifi, Coastal Kenya, over a 12-Year period, 2007–2018. Open. Forum Infect. Dis.8 (12), ofab571. https://doi.org/10.1093/ofid/ofab571 (2021).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol.30 (4), 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol.32 (1), 268–274. https://doi.org/10.1093/molbev/msu300 (2015).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol.2 (1). https://doi.org/10.1093/ve/vew007 (2016).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T. Y. Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol.8 (1), 28–36. https://doi.org/10.1111/2041-210X.12628 (2017).
R Core Team. R: A language and environment for statistical computing. Published online 2013. Accessed November 9. URL (2021). http://www.R-project.org/
Spellerberg, I. F. & Fedor, P. J. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon–Wiener’ Index. Glob. Ecol. Biogeogr.12 (3), 177–179. https://doi.org/10.1046/j.1466-822X.2003.00015.x (2003).
SIMPSON EH. Measurement of Diversity. Nature. 163 (4148), 688. https://doi.org/10.1038/163688a0 (1949).
Milanoi, S., Ongus, J. R., Gachara, G., Coldren, R. & Bulimo, W. Serotype and genetic diversity of human rhinovirus strains that circulated in Kenya in 2008. Influenza Other Respir Viruses. 10 (3), 185–191. https://doi.org/10.1111/irv.12373 (2016).
Briese, T. et al. Global distribution of novel rhinovirus genotype. Emerg. Infect. Dis.14 (6), 944–947. https://doi.org/10.3201/eid1406.080271 (2008).
Sansone, M. et al. Rhinovirus infections in western Sweden: a four-year molecular epidemiology study comparing local and globally appearing types. Eur. J. Clin. Microbiol. Infect. Dis.32 (7), 947–954. https://doi.org/10.1007/s10096-013-1832-x (2013).
Luka, M. M. et al. Rhinovirus dynamics across different social structures. npj Viruses. 1 (1), 6. https://doi.org/10.1038/s44298-023-00008-y (2023).
Monto, A. S. Epidemiology of viral respiratory infections. Am. J. Med.112(Suppl), 4S-12S. https://doi.org/10.1016/s0002-9343(01)01058-0 (2002).
Arden, K. E., McErlean, P., Nissen, M. D., Sloots, T. P. & Mackay, I. M. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J. Med. Virol.78 (9), 1232–1240. https://doi.org/10.1002/jmv.20689 (2006).
Luka, M. M. et al. Whole genome sequencing of two human rhinovirus a types (A101 and A15) detected in Kenya, 2016–2018. Wellcome Open. Res. 2021. 6, 178. https://doi.org/10.12688/wellcomeopenres.16911.1 (2021).
World Health Organisation. Pocket Book of Hospital Care for Children: Guidelines for the Management of Common Childhood Illnesses. ;(2nd edition.). (2013).
Acknowledgements
We thank all the study participants for their contribution of study specimens and data and surveillance staff from all the study sites included. We acknowledge the laboratory staff at KEMRI-Wellcome Trust Research Programme and CDC-supported laboratory at KEMRI-Centre for Global Health Research for specimen handling and testing. The primary data and specimen collection in Influenza surveillance and PBIDS sites were supported by U.S. Centers for Disease Control and Prevention while pediatric surveillance at Kilifi County Hospital was funded by the Wellcome Trust.
Funding
This work was supported by Wellcome [102975], and U.S. Centers for Disease Control and Prevention through Cooperative Agreement [#6U01GH002143].
Author information
Authors and Affiliations
Contributions
J.M.M, E.K., C.A., PKM, D.J.N., Conceptualised and designed the studyJ.M.M.,M.M.L, C.L., M.M., Provided laboratory sequencing and support J.M.M., M.M.L, E.K., C.A., D.J.N., Provided phylogenetic analysis J.M.M., N.M. Provided data curation and analysisJ.M.M., E.K., C.A., D.J.N., PKM Wrote the original manuscript draftAll authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Disclaimer
The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of U.S. the Centers for Disease Control and Prevention.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Morobe, J.M., Kamau, E., Luka, M.M. et al. Spatio-temporal distribution of rhinovirus types in Kenya: a retrospective analysis, 2014. Sci Rep 14, 22298 (2024). https://doi.org/10.1038/s41598-024-73856-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-73856-0
