
Abstract
The small genera Allaeanthus and Malaisia within the Moraceae have important edible, medicinal, and economic value. However, complete plastome blueprints and a well-resolved evolutionary history of these two genera are still lack, thereby limiting their conservation and application. The recent discovery of a new distribution of Allaeanthus kurzii in Hainan, China, marked by the collection of two unique samples, alongside three samples of Malaisia scandens, has opened new avenues for research. This study aimed to compare the Allaeanthus and Malaisia plastomes of Hainan Province samples with those of samples from other regions, focusing on plastome structure, codon usage bias, natural selection, and the evolutionary history of A. kurzii and M. scandens. The results showed that both species had a quadripartite plastome structure, with sizes ranging from 162,134 to 162,170 bp for A. kurzii and 161,235 to 162,134 bp for M. scandens. Both species displayed loss of the infA gene and reduction of the rpl22 gene. Two highly variable regions (petD-trnD-GUC and rpl20-clpP) and three highly variable genes (rpl20, petB, and rpl16) were identified in A. kurzii, while two highly variable regions (ycf2-ndhB and ccsA-ndhE) and three highly variable genes (psbT, rpl36, and ycf2) were found in M. scandens. The protein-coding sequences (CDSs) of the Allaeanthus and Malaisia plastomes exhibited similar patterns of adaptive indices and codon usage frequencies. The genes associated with photosynthesis underwent strong purifying selection. Phylogenetic analysis revealed that Allaeanthus, Broussonetia, and Malaisia constituted a monophyletic group, with Malaisia being more closely related to Broussonetia. Broussonetia diversified approximately 19.78 million years ago, Malaisia approximately 4.74 million years ago, and Allaeanthus approximately 16.18 million years ago. These new plastome-based discoveries will guide conservation planners and medicinal plant breeders and genetic resource development for these species in the region.
Similar content being viewed by others

Introduction
The Moraceae family consists of 7 tribes, 48 genera, and approximately 1200 species1,2,3. Within this family, Allaeanthus Thwaites and Malaisia Blanco are two small genera from the tribe Dorstenieae. Allaeanthus comprises four species, whereas Malaisia is represented by a single species, Malaisia scandens (Lour.) Planch. These plants hold significant economic value and are deeply rooted in cultural history. Allaeanthus kurzii Hook. f. has edible leaves and fruits with a sweet taste4, while its bark was utilized for papermaking and fiber production5. The branches of A. kurzii are also used in traditional medicine to treat conditions such as rheumatism. On the other hand, M. scandens had strong and tough stem bark fibers suitable for making ropes, with its roots and leaves used medicinally for various purposes. Both genera are predominantly found in tropical or subtropical regions of Asia. Allaeanthus kurzii is mainly distributed in China, Vietnam, Laos, Thailand, Myanmar, Bhutan, and India, while M. scandens is found in China, Southeast Asia, and the western Pacific islands6. This research marked the first identification of A. kurzii distribution in Hainan, China, offering crucial insights for future studies on the ecology and conservation of this species.
Allaeanthus was initially described by Thwaites (1854)5, and then Corner (1962)7 classified Broussonetia L’Hér. ex Vent. into sect. Broussonetia and sect. Allaeanthus, but there were no significant differences between them. These three genera include dioecious plants, trees, or climbing plants with deciduous leaves, alternate or (nearly) opposite leaves, spike-like male inflorescences (sometimes head-shaped), and head-shaped or nearly spherical female inflorescences8. Pollen morphological studies by Kim et al. (1993)9 revealed that the pollen morphology of sect. Allaeanthus and Malaisia closely resembled that of species in sect. Broussonetia, making differentiation challenging. Subsequent molecular studies contradicted past morphology-based classifications. Chung et al. (2017)2 utilized the ndhF gene from the plastome and the 26S subunit sequence from the nuclear genome to show that sects. Broussonetia and Allaeanthus were monophyletic groups. However, based on Corner’s (1962) classification criteria2, Broussonetia was considered polyphyletic, leading to the reinstatement of sect. Allaeanthus as a separate genus for maintaining monophyly. Berg (1962)7 previously placed M. scandens in the genus Trophis P. Browne. Recent molecular research indicated that Broussonetia and M. scandens are part of the same clade, establishing Trophis as monophyletic2,10,11,12. Malaisia has been reinstated as an independent genus, alongside Broussonetia and Allaeanthus. There are three species of Broussonetia (B. kaempferi Siebold, B. monoica Hance, and B. papyrifera (L.) L’Hér. ex Vent.), as well as one hybrid species (B. × kazinoki Siebold)8. Allaeanthus comprises four species: A. greveana (Baillon) C.C.Berg, A. zeylanica (Thwaites) Corner, A. kurzii, and A. luzonicus (Blanco) Bureau2. Malaisia, on the other hand, is represented by a single species, Malaisia scandens8. In a word, despite the limited number of species within these genera, there has been significant debate regarding their phylogenetic positions.
In angiosperms, plastids, which include chloroplasts, chromoplasts, and leucoplasts, are crucial organelles in plant cells for synthetic metabolism. Plastids have independent genetic information and can transcribe and translate autonomously. The plastome consists of a large single-copy region (LSC), a small single-copy region (SSC), and a pair of inverted repeats (IRs), which typically range in size from 120 to 160 bp13,14. The plastome is smaller, maternally inherited, structurally conserved with rare recombination events, exhibits moderate nucleotide substitution rates, and shows significant differences in evolutionary rates across different regions. This makes it advantageous for phylogenetic studies at both the species and higher taxonomic levels15,16,17,18,19.
For Allaeanthus and Malaisia, a small number of plastomes have been used to investigate the phylogenetic relationships of some specific species in particular regions8. However, more plastome data of these two genera are required for developing more universal molecular markers for identifying and screening Allaeanthus and Malaisia species. In addition, an investigation incorporating plastomes of additional Allaeanthus and Malaisia samples would provide us a more comprehensive view to the phylogenetic relationships and evolutionary history within these two genera, which serves as a foundation for the correct utilization and development of Allaeanthus and Malaisia species. This study revealed a new distribution of A. kurzii in Hainan, China, with two samples successfully collected. Its morphology is shown in Fig. 1. Additionally, three samples of M. scandens from Hainan, China, were also collected (Fig. S1). All five complete plastomes were sequenced and characterized. The specific objectives of this study were: (1) to compare the differences at the plastome level between A. kurzii and M. scandens species in Hainan, China, and those from other regions and (2) to validate the phylogenetic relationships among Broussonetia, Allaeanthus, and Malaisia, and estimate their divergence times. These findings will offer valuable insights for resource utilization and conservation efforts for these species.

Morphology of Allaeanthus kurzii (Photos from Lang-Xing Yuan): (a) Allaeanthus kurzii plant. (b) A detailed diagram of a staminate inflorescence. (c) Staminate inflorescences. (d) A detailed diagram of a pistillate inflorescence. (e) Stem and leaf petiole. (f) Complete leaf, including both the upper (adaxial) and lower (abaxial) surfaces. (g) Pistillate inflorescence.
Results
Characteristics of the A. kurzii and M. scandens plastomes
The plastome structures of A. kurzii (162,134 bp–162,140 bp) and M. scandens (161,748 bp–162,134 bp) were relatively conserved, displaying a classic tetrad structure with a large single copy region (LSC; 90,154 bp–90,160 bp, 89,722 bp–89,828 bp, respectively), a small single copy region (SSC; 20,146 bp–20,146 bp, 19,873 bp–20,037 bp, respectively), and a pair of inverted repeat regions (IRa and IRb; 25,917 bp–25,917 bp, 25,820 bp–26,177 bp, respectively) (Fig. 2). The plastome structures of the three A. kurzii samples showed a high level of consistency (Fig. 2a, Table S3). In contrast, the plastome structures of M. scandens exhibited some differences among the different samples, particularly in terms of total length, especially in the IR and SSC regions of the samples collected from Hainan, China (Fig. 2b). Variations in region length were also observed when comparing the M. scandens sample from Taiwan, China (Table S2). The plastomes of the A. kurzii and M. scandens samples were annotated with 136–137 genes, including protein-coding genes, tRNA genes, rRNA genes, and pseudogenes (Tables S2, S3). The difference in gene numbers between the two species was primarily due to a ψrps19 pseudogene in A. kurzii compared to M. scandens. In A. kurzii, the rps19 gene spanned the JLB boundary, resulting in the presence of the pseudogene ψrps19 in the IRa region (Fig. 3).

Plastome maps of A. kurzii and M. scandens.(a) The gene map of the A. kurzii plastomes; (b) the gene map of the M. scandens plastomes. From the center outward, the first track shows forward and reverse repeats connected by red and green arcs. The second-track concatenated repeats are displayed as short blue bars. The third track also displays microsatellite sequences as short green and yellow bars. The line graph on the fourth track shows the frequency of alternate bases at specific locations. The frequencies of bases ‘A’, ‘G’, ‘C’, and ‘T’ are represented by red, blue, orange, and green lines, respectively. The length of the lines represents the frequency of substitution. The fifth track displays the regions of small single copy (SSC), inverted repeats (IRa and IRb), and large single copy (LSC) in the genome. The sixth track displays the GC content of the genome. The seventh track displays the genes. Optional codons are indicated by deviations in parentheses following the gene names. Genes are color-coded by their functional categories. The transcription directions of internal and external genes are clockwise and counterclockwise, respectively.

The expansion and contraction of the IR boundaries were compared in the plastomes of these seven A. kurzii and M. scandens samples.
The analysis of three A. kurzii samples revealed consistent GC content across different regions, contrasting with the minimal GC content variation observed in the plastomes of M. scandens. Specifically, the MSC2 sample of M. scandens displayed slightly lower GC content in the SSC region (28.8% compared to 28.9%) and the IR region (42.7% compared to 42.8%) than the other samples. Overall, both plant species exhibited a stable GC content with heightened conservation in their plastomes, showcasing a relatively high GC content in the IR region and a relatively low GC content in the SSC region (Table S4).
Nucleotide diversity analysis
After conducting sliding window analysis using DnaSP v5.0, nucleotide polymorphism (Pi) was calculated for the plastome regions (LSC, SSC, and IR) of A. kurzii and M. scandens. The Pi values for A. kurzii ranged from 0 to 3.33 × 10−3, with the petD-trnD-GUC and rpl20-clpP regions in the LSC region showing the same level of variation at 3.33 × 10−3 (Fig. 4a). In contrast, the Pi values for M. scandens ranged from 0 to 4.66 × 10−2, with the ycf2-ndhB region displaying the greatest variation at 4.66 × 10−2, and the ccsA-ndhE region showing relatively less variation at 1.61 × 10−2 (Fig. 4c). Further analysis of 80 shared protein-coding genes related to nucleotide polymorphism (Pi) revealed that most genes in A. kurzii were highly conserved, with only rpl20, petB, and rpl16 showing relatively high variability (Fig. 4b). Similarly, in M. scandens, most genes were highly conserved, except for psbT, rpl36, and ycf2, which exhibited greater variability (Fig. 4d). Overall, the LSC and SSC regions showed greater variability than did the IR region, and non-protein-coding regions displayed greater variability than did protein-coding regions.

Sliding window analysis of the plastomes was performed on three samples of A. kurzii and four samples of M. scandens. (a) The complete plastomes of three A. kurzii samples were subjected to sliding window analysis. (b) Nucleotide polymorphism analysis of protein-coding genes was conducted for three A. kurzii samples. (c) The complete plastomes of four M. scandens samples were analyzed using sliding window analysis. (d) Nucleotide polymorphism analysis of protein-coding genes was performed on four M. scandens samples. The sliding window analysis was carried out with a window length of 600 bp and a step size of 200 bp. The dashed lines indicate Pi values of 0.0025 in (a), 0.0015 in (b), 0.015 in (c), and 0.004 in (d).
Repetitive sequences in the plastomes
The distribution of repeat sequences in the plastomes of two A. kurzii samples from Hainan, China, revealed 17 forward repeats, 25 palindromic repeats, 3 reverse repeats, and 1 complementary repeat. A comparison with the Thailand samples revealed a slight increase in the number of detected repeat sequences: 20 forward repeats (an increase of 3) and 26 palindromic repeats (an increase of 1). In the case of the plant species M. scandens, the MSC1 sample (located in Liangyuan, Danzhou city, Hainan Province) exhibited the greatest number of forward repeat sequences (28 pairs), while the MSC2 sample (located in Daguangba, Dongfang city) had the fewest (20 pairs). The range of reverse repeat sequences was 3–12 pairs, with the MSC1 sample showing the most (12 pairs), three times more than other samples with only 3 or 4 pairs. Among the complementary repeat sequences, the MSC2 sample had the most (5 pairs), followed by the MSC3 sample (located in Tongcai village, Wangxiatown, Changjiang Li) with 2 pairs, while the other samples had 1 pair each. The range of palindrome repeat sequences was 20–22 pairs, except for the MSC2 sample, which had 22 pairs, while the rest had 20 pairs. The Thailand sample exhibited 30 forward repeat sequences, 3 reverse repeat sequences, 1 complementary repeat sequence, and 20 palindrome repeat sequences (Table S5).
SSR analysis was performed on the plastomes of seven samples, revealing that mononucleotide repeat sequences were the most prevalent, accounting for 61.38% of the total repeats. The A. kurzii samples exhibited more SSR repeats than did the M. scandens samples, except for pentanucleotide repeats. The majority of SSR loci were found in intergenic regions of the plastome (65.93%), with a smaller proportion in protein-coding regions (34.06%). Statistical analysis indicated that SSR loci were predominantly concentrated in the LSC region (60–83), followed by the SSC region (21–24), and few were present in the IR region (2–6) (Figs. 5, 6, Table S5).

Statistics of repeat sequences in each sample of seven A. kurzii and M. scandens samples. (a) Overall count of tandem repeat sequences and dispersed repeat sequences in each sample. (b) Distribution of six types of simple repeat sequences in each sample. (c) Distribution of simple repeat sequences in coding and noncoding regions. (d) Distribution of simple repeat sequences in the LSC, IR, and SSC regions.

Statistics on the proportion of various types of information in repeated sequences in each sample of seven A. kurzii and M. scandens samples. (a) The proportions of tandem and dispersed repeat sequences were calculated. (b) The percentage of simple repeat sequences in protein-coding regions (CDS) and noncoding regions (IGS) was determined. (c) The proportions of the six types of simple repeat sequences ware analyzed. (d) The proportions of simple repeat sequences in the LSC region, IR region, and SSC region.
Codon usage bias
This study screened 52 common protein-coding genes in A. kurzii and M. scandens. Codon preference analysis based on species showed that leucine (Leu) was the most frequently used amino acid in both species, representing 10.75 and 10.68% of all amino acids, with 6806 and 8995 corresponding codons, respectively. Additionally, isoleucine (Ile) had AUU as the predominant codon in both species, accounting for 4.30 and 4.25%, respectively. Notably, cysteine (Cys) had the lowest frequency of usage, accounting for only 1.08% and 1.07% of all amino acids in the plastomes of A. kurzii and M. scandens, respectively. The codon UGC encoding cysteine had the lowest frequency of usage in both species, at 0.25 and 0.26%, respectively (Fig. 7). Analysis of the RSCU values for protein-coding genes in the plastomes of A. kurzii and M. scandens revealed 30 codons with RSCU values above 1, indicating a greater frequency of usage, and 34 codons with RSCU values equal to or less than 1, indicating a lower frequency of usage. Further investigation revealed that high-frequency synonymous codons ended with A and U, while low-frequency synonymous codons ended with C and G (Fig. 7).

The RSCU analysis was conducted on seven A. kurzii and M. scandens samples. (a) RSCU analysis was performed on three A. kurzii samples. (b) RSCU analysis was conducted on four M. scandens samples. The x-axis represents amino acids, with the corresponding codons encoding the amino acids below. The y-axis represents the RSCU usage frequency of each codon.
Codon analysis revealed a bias toward 0 in the CAI values, indicating a weaker adaptability of genes to optimal codons. Similarly, the CBI values showed a bias toward -1, suggesting a lower preference of amino acids for specific codons. The Fop values for the two species were 0.350 and 0.351, respectively, indicating a relatively low frequency of optimal codon usage. The ENC values ranged from 48.46 to 48.60, showing high consistency among different samples and species, indicating a relatively weak preference for codon usage. The GC3s contents of A. kurzii and M. scandens did not significantly differ among the samples, with both species displaying a consistent pattern of optimal codon usage. The ENC-plot was used to measure the degree of synonymous codon usage bias and deviation from random selection, with GC3s and ENC as the x and y coordinates, respectively. Overall, A. kurzii and M. scandens showed similar patterns in codon usage preference, with most genes favoring certain codons primarily due to mutation, while only a few genes were influenced by natural selection. The PR2-plot analysis focused on analyzing the preference of codons for the four bases, A, T, G, and C, at the third position in genes. The results for A. kurzii and M. scandens indicated a preference for the third base in gene codons, specifically T > A and G > C. Additionally, specific genes (rpl16, rps14, psbD, and psbA) in both species exhibited abnormal patterns, with differences in base content such as increased G content in the rps4 gene, increased AG content in the rpl16 gene, increased TC content in the psbA gene, and a tendency toward increased TC content in the psbD gene (Fig. 8).

ENC-plot and PR2-plot analyses were conducted on protein-coding genes in three A. kurzii and four M. scandens plastomes. (a) ENC-plot analysis was performed on the three A. kurzii samples, with solid lines representing the expected values and data points indicating the observed values. (b) A detailed analysis was conducted on specific genes in the three A. kurzii samples that deviated from the expected values. (c) The PR2-plot analysis was carried out on the three A. kurzii samples. (d) ENC plot analysis was performed on the four M. scandens samples, with solid lines representing the expected values and data points indicating the observed values. (e) A detailed analysis was conducted on specific genes in the four M. scandens samples that deviated from the expected values. (f) PR2-plot analysis was carried out on the four M. scandens samples.
Phylogenetic relationships based on the plastomes
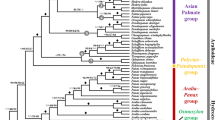
Using two datasets, our phylogenetic study conducted analyses on sequence matrices. The first dataset included large single-copy regions (LSC), small single-copy regions (SSC), and a single inverted repeat region (IR). The aligned matrix had a total length of 151,653 base pairs (bp), with 13,543 parsimony-informative sites, 14,320 singleton sites, and 123,790 constant sites identified. The second dataset comprised a matrix of plastid protein-coding genes without duplicates, with an alignment length of 70,974 bp, 4,601 parsimony-informative sites, 4,797 singleton sites, and 61,575 constant sites. Both Bayesian Inference (BI) and Maximum Likelihood (ML) analyses demonstrated no differences in the topology and support values of the phylogenetic trees constructed from these two datasets. Full support was observed for all nodes in the phylogenetic tree. Notably, our study revealed that samples of A. kurzii from Hainan, China, clustered with a sample from Thailand, while samples of M. scandens from Hainan, China, grouped with a sample from Taiwan, China (Fig. 9). In summary, phylogenetic analysis revealed that Allaeanthus, Broussonetia, and Malaisia constituted a monophyletic group respectively, with Malaisia being more closely related to Broussonetia (Fig. 9).

A phylogenetic tree based on the complete plastome sequences of 23 Moraceae species was constructed and analyzed using maximum likelihood (ML) and Bayesian Inference (BI).
Evolution rate analysis (dN/dS)
To further explore the adaptive evolutionary mechanisms of the A. kurzii and M. scandens genes, we analyzed the dN/dS values for 80 shared plastid protein-coding genes. Compared to Antiaris toxicaria Lesch. As a reference, only the self-replicating type rpl2 gene in M. scandens exhibited positive selection (dN/dS value = 1.27), while other genes showed dN/dS values below 1, indicating of purifying selection. In A. kurzii, positive selection was observed for the self-replicating type rpl22 gene (dN/dS value = 1.32), with other genes also under purifying selection (Fig. 10c). When Ficus maxima Mill. was used as the reference, both A. kurzii and M. scandens displayed positive selection in the rpl22 gene. Additionally, A. kurzii showed positive selection in the psbH gene. The dN/dS values for the remaining genes were less than 1, suggesting purifying selection (Fig. 10d).

Analyses of evolutionary pressure on plastid gene homologues in the A. kurzii and M. scandens. (a) The dN/dS ratios were compared between three samples of A. kurzii and four samples of M. scandens for genes related to photosynthesis, self-replication, and other protein-coding genes, using A. toxicaria as the reference. (b) The dN/dS ratios were compared between three samples of A. kurzii and four samples of M. scandens samples for genes related to photosynthesis, self-replication, and other protein-coding genes, using F. maxima as the reference. (c) The dN/dS values were calculated for three samples of A. kurzii and four samples of M. scandens, using A. toxicaria as the reference. (d) The dN/dS values were calculated for three samples of A. kurzii and four samples of M. scandens, using F. maxima as the reference. The green line represents A. kurzii, the blue line represents M. scandens, and the black dashed line represents dN/dS = 1.
The selected genes were categorized into photosynthetic, self-replicating, and other types. Comparison of the dN/dS values of various functional genes between A. kurzii and M. scandens revealed no significant differences in these three gene categories between the two species. Additionally, the analysis indicated notable differences in selection pressure between photosynthetic genes and other types within both M. scandens and A. kurzii (Fig. 10a,b).
Estimation of divergence time
The BEAST-derived chronogram of Moraceae (Fig. 11), based on the CDSs database, indicated a posterior probability of 1 for all nodes, suggesting that the crown group node of Moraceae occurred approximately 85.40 million years ago (95% HPD: 78.87–91.34 Ma). The divergence time of node B was estimated to be approximately 74.93 million years ago (95% HPD: 72.98–76.88 Ma). Nodes D and E show the divergence of Broussonetia, Malaisia, Allaeanthus, Ficus L., Olmedia Ruiz & Pav., and Antiaris Lesch. genera approximately 42.53 million years ago (95% HPD: 33.69–60.25 Ma) and 55.98 million years ago (95% HPD: 54.02–57.93 Ma), respectively. The crown group node of Broussonetia was placed in the early Pleistocene approximately 19.78 million years ago (95% HPD: 8.07–32.55 Ma). Allaeanthus differentiated approximately 16.18 million years ago (95% HPD: 4.53–33.12 Ma), while M. scandens emerged approximately 4.74 million years ago (95% HPD: 0.86–13.59 Ma). Notably, samples from Hainan, China, showed relatively recent divergence times for M. scandens and A. kurzii. Node C, comprising Morus L. and Artocarpus J. R. Forst. & G. Forst. was estimated to have diverged from the late Cretaceous to early Tertiary approximately 72.14 million years ago (95% HPD: 70.30–74.05 Ma) (Table 1).

BEAST-derived chronograms of Moraceae based on CDSs of the plastome, with nodes A, B, C, D, and E further explained in Table 1.
Discussion
Plant diversity surveys conducted in the Hainan region led to the first discovery of the distribution of A. kurzii in this area, providing valuable evidence for further research on its distribution and ecological characteristics. The plastomes of the two A. kurzii samples exhibited a typical quadripartite structure, with genome lengths of 162,134 and 162,140 bp, respectively, and a GC content of approximately 35.7%. Both samples contained 137 genes. A comparison with A. kurzii sample from Thailand revealed differences in plastid protein-coding gene length, while the plastome gene content and structure were more consistent. Additionally, three plastomes of M. scandens from Hainan were assembled, each containing 136 genes, with variations in plastome structures among them and differences compared to samples from Taiwan. Both M. scandens and A. kurzii plastomes displayed conserved GC content, with higher AT content than GC content. Sequence alignments showed no significant rearrangements and high sequence homology among them. Comparing plastomes from different collection sites revealed that differences in plastome composition increased with the distance between collection points, possibly due to natural selection, environmental adaptability, and genetic diversity.
The gene content, number, and structure in the plastomes of most autotrophic land plants are typically conserved20,21. However, certain plastid protein-coding genes can be lost in specific species, such as rpl22 in Fabaceae22 and infA in Solanum23, indicating the common phenomenon of gene transfer and loss in plant plastomes. The absence of the infA gene has also been observed in M. scandens and A. kurzii. Previous studies have demonstrated that infA plays a crucial role in Escherichia coli (Migula) Castellani & Chalmers24, working with two nucleus-encoded initiation factors to initiate translation and facilitate interactions between mRNA, ribosomes, and the initiator tRNA-Met23. This gene has undergone multiple losses or transfers during the evolutionary history of angiosperms23. It has been established that a nucleus-encoded infA gene is present in various species such as Arabidopsis thaliana (L.) Heynh., Glycine max (L.) Merr., Solanum lycopersicum L., and Mesembryanthemum crystallinum L., where it is translated in the cytoplasm before being transported into the plastids23. The infA gene is considered one of the most mobile plastid genes in plants due to its recurrent independent losses and transfers in plastomes23. Meanwhile, compared with the plastome of other Moraceae species, it was observed that the rpl22 gene was missing in B. kaempteri and B. monoica, consistent with previous findings8. Additionally, a reduction in the size of the rpl22 gene was noted in certain samples of A. kurzii, M. scandens, and A. luzonicus plastomes, with dN/dS values suggesting strong positive selection on the rpl22 gene. No reduction or loss of the rpl22 gene was seen in other Moraceae species’ plastomes examined. This discrepancy may be attributed to a historical event where the common ancestor of Broussonetia, Malaisia, and Allaeanthus experienced rpl22 gene degradation, resulting in complete loss in B. kaempteri and B. monoica, but restoration in B. papyrifera. Further investigation is warranted to explore this phenomenon. Furthermore, it was found that the rpl22 gene in M. scandens and A. luzonicus plastomes from different locations underwent contraction, suggesting this phenomenon is not location dependent. Research has shown that rpl22 gene loss in angiosperm plastomes is relatively common, with examples of transfer to the nuclear genome. In land plants, plastid gene loss can occur through transfer to the nucleus, substitution with nucleus-encoded mitochondrial targeting genes, or nuclear gene replacement of plastid genes. In the Moraceae family, infA and rpl22 genes were transferred to the nucleus, while no loss of other genes was observed.
The plastome of M. scandens had one less gene than that of A. kurzii due to the expansion and contraction of the inverted repeat (IR) regions within the plastome25,26. In A. kurzii, a part of the rps19 gene expanded into the IRb region, leading to a pseudogene ψrps19 in the IRa region, which is commonly observed in angiosperms14. Additionally, it was discovered that the dN/dS values of photosynthesis-related genes were notably lower than those of self-replication-related genes and other genes in both M. scandens and A. kurzii, indicating strong purifying selection on photosynthesis-related genes. This implies that natural selection is promoting the optimization of photosynthesis-related genes, preserving advantageous variations for photosynthesis within the species27.
Previous controversies surrounding the classification of Broussonetia, Allaeanthus, and Malaisia have been addressed through various studies. Initial findings in 1933 suggested similarities in pollen morphology between certain species of Allaeanthus and Malaisia with Broussonetia9, aligning with Corner’s classification7. However, subsequent research utilizing plastomes and nuclear fragments argued for the reclassification of M. scandens as a distinct Malaisia species and the reinstatement of Sect. Allaeanthus as an independent genus2,8. Our research supports this reclassification, highlighting the monophyletic grouping of M. scandens samples, A. kurzii and A. luzonicus samples, respectively. This emphasizes the limitations of relying solely on morphological characteristics for classification and underscores the importance of plastome studies. Recent advancements in utilizing hotspot mutations as genetic markers have proven valuable in taxonomic studies28, including our analysis of Broussonetia, Allaeanthus, and Malaisia. Our comprehensive plastome analysis identified highly variable regions (petD-trnD-GUC and rpl20-clpP in A. kurzii; ycf2-ndhB and ccsA-ndhE in M. scandens) and genes (rpl20, petB, and rpl16 in A. kurzii; psbT, rpl36, and ycf2 in M. scandens), which could serve as potential molecular markers for further species studies. Through comparative studies of samples from Malaisia and Allaeanthus, it was observed that the differentiation of M. scandens individuals in Hainan, China occurred slightly later than those in Taiwan, China. Similarly, the differentiation of A. kurzii samples in Hainan, China, occurred slightly later than in Thailand. These variations could be attributed to factors such as geographical isolation. Moreover, environmental factors like climate, temperature, humidity, and soil type may differ across regions, prompting species to adapt to various ecological environments. The estimated crown age of the Moraceae was approximately 78.87–91.34 million years ago, aligning closely with previous research, such as studies of Zetega et al.29 (around 72.60–110.00 million years ago) and Qian et al.3 (approximately 73.30–84.70 million years ago).
Codon usage preference is a common phenomenon, which is influenced by mutation bias, selection, or a combination30. The similar codon usage preference patterns seen in A. kurzii and M. scandens may be due to similar selective pressures and ecological environments during their evolution. Selection pressure plays a role in shaping codon preference, with both species primarily found in tropical or subtropical regions of Asia8, suggesting they face similar selective pressures. Similar habitats can result in similar nutritional requirements, metabolic pathways, and protein functional demands, leading to similar codon usage characteristics. Additionally, gene expression levels and transcriptional regulation may also influence codon preference31, although gene expression is mainly influenced by transcription and post-transcriptional regulation32. ENC analysis revealed significant deviation from expected values in rps8, rps14, psbA, ycf2, petA, and ndhF in both species, indicating consistency. Highly expressed genes tend to have stronger codon preference, encoding optimal codons corresponding to abundant tRNA for faster and more efficient translation. This suggests similarities in protein-coding gene expression levels and regulatory mechanisms, leading to a tendency for similar codon preference. The GC content of a genome is influenced by the mutation process and plays a key role in codon variation between species32,33. In the coding sequences of A. kurzii and M. scandens, the third position of codons is mainly composed of A/T, with a preference for codons ending in A/U, which aligns with angiosperm characteristics. Analysis of the effective number of codons (ENC) showed that most genes had ENC values close to the expected curve, while a few genes exhibited significantly lower ENC values, indicating the impact of natural selection on these specific genes. Notably, genes affected by natural selection in both A. kurzii and M. scandens include rps8, rps14, psbA, ycf2, petA, ndhF, ndhI, and ndhC in A. kurzii, as well as rps8, rps14, psbA, ycf2, petA, ndhF, rps18, and rpl16 in M. scandens. These results suggest that codon usage preferences in A. kurzii and M. scandens are influenced by both mutation and natural selection. Furthermore, they support the idea that the GC content of the genome is determined by the mutation process and is a significant factor in codon usage variation between species, consistent with previous research findings34,35.
Conclusions
The present study has provided valuable insights into the plastome characteristics and evolutionary history of the small but significant Allaeanthus and Malaisia within the Moraceae family. The identification of new distribution records for Allaeanthus kurzii in Hainan, China, and the subsequent comparative analysis of Allaeanthus and Malaisia plastomes with samples from other regions, have yielded important findings. All plastomes exhibited a conserved quadripartite plastome structure, with notable gene loss and reduction events, suggesting a shared evolutionary trajectory. The patterns of adaptive indices and codon usage frequencies, along with the strong purifying selection observed in photosynthesis-related genes, indicate functional constraints and potential adaptations to their environments. The identification of highly variable regions and genes in both genera provides potential markers for future genetic and phylogenetic studies. Phylogenetic analysis offers a clearer understanding of their evolutionary history and biogeography.
Methods
Sample collection and DNA extraction
Five wild samples were collected from Hainan Province, China, including two newly recorded samples of A. kurzii and three samples of M. scandens (Table S1). The experimental numbers for the two samples of A. kurzii are MSN1 (Voucher specimen number: Pan Li 010350) and MSN2 (Voucher specimen number: Pan Li 010351), while the experimental numbers for the three samples of M. scandens are MSC1 (Voucher specimen number: Pan Li 010352), MSC2 (Voucher specimen number: Pan Li 010353), and MSC3 (Voucher specimen number: Pan Li 010354). All samples were identified by the second author (Lang-Xing Yuan) and the third author (Pan Li). Silica-dried leaves from these two species were used for DNA extraction. Genomic DNA was extracted using a modified CTAB protocol36. The voucher specimens of the aforementioned samples are deposited in the Specimen Museum of Wenzhou University(WZU). As the species A. kurzii and M. scandens were not listed in the “Threatened Species List of China’s Higher Plants37” and were not collected from protected areas, sampling did not require permission. All experiments, including the collection of plant materials, complied with relevant institutional, national, and international guidelines and legislation. Plastome sequences of A. kurzii (NC_041637)2 from Thailand and M. scandens (NC_047182)2 from Taiwan were obtained from the NCBI database. Plastome sequences of 15 species from five tribes in the family Moraceae, along with two outgroup species Boehmeria nivea (L.) Gaudich. and Cannabis sativa L. (Table S1), were also downloaded for phylogenetic analysis.
Genome sequencing, assembly, and annotation
The extracted DNA was quantified to ensure accurate concentration levels for sequencing and then sent to the Beijing Genomics Institute (Shenzhen, China) for plastome sequencing using the BGISEQ platform. This platform is known for its high-throughput sequencing capabilities, making it suitable for comprehensive plastome analysis.The raw data were processed using Fastp v0.12.438, a tool designed to filter out reads containing adapters, remove low-quality sequences, and trim the data for better accuracy. Additionally, the data quality was assessed using Fastqc software which provides detailed reports on sequence quality, ensuring that only high-quality Clean Data were used for further analysis. The organelle genomes were assembled by the GetOrganelle pipeline39, a specialized tool for de novo assembly of organelle genomes, which efficiently constructs complete and accurate plastomes by leveraging the unique characteristics of organelle DNA. Following assembly, the plastomes were annotated using the online tool CPGAVAS2 (http://www.herbalgenomics.org/cpgavas), an online tool specifically designed for annotating chloroplast genomes, and manual adjustments were performed using Geneious Prime 2021 to eliminate errors and redundant annotations.
Finally, the plastome structure and gene content were visualized using OGDRAW v1.3.140 (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html), a tool that generates detailed graphical representations of organelle genomes, helping to illustrate the overall structure and gene arrangement within the plastomes. This visualization step is crucial for confirming the integrity of the assembly and for facilitating comparative genomics studies.
Comparative genomic analysis
The analysis of plastome expansion and contraction in the inverted repeat (IR) region, as well as polymorphisms in the boundary region, was performed using the IRscope script41. This tool is specifically designed to visualize and compare the boundaries of IR regions across different plastomes, helping to identify any expansion or contraction events that may have occurred. To further analyze and compare the plastomes of the A. kurzii and M. scandens, the sequences were aligned using the LAGAN model42, a tool available on the mVISTA website43. The LAGAN model is particularly effective for aligning large sequences, such as plastomes, and the mVISTA platform provides a user-friendly interface for visualizing these alignments, allowing researchers to easily detect conserved and divergent regions.
Nucleotide polymorphism analysis of the plastomes of A. kurzii and M. scandens was performed using DnaSP v5.0 software44. This software allows for the calculation of various measures of nucleotide diversity, providing insights into the genetic variation within and between the plastomes of A. kurzii and M. scandens. For this analysis, the window length was set to 600 bp, with a step size of 200 bp, enabling a detailed examination of polymorphism patterns across the plastomes.
Repeat sequence analysis
The REPuter tool45 (https://bibiserv.cebitec.uni-bielefeld.de/reputer) was utilized to search for dispersed repeat sequences in all plastomes. This tool specializes in identifying different types of repeats, including forward repeats (F), reverse repeats (R), complementary repeats (C), and palindromic repeats (P). For this analysis, the minimum repeat sequence size was set to ≥ 30 bp, allowing for the detection of significant repeat sequences, with a maximum calculated repeat count of 80 and a Hamming distance of 3, which ensures tolerance for small mismatches in the repeat sequences. Tandem Repeats Finder46 (https://tandem.bu.edu/trf/trf.html) was utilized to identify tandem repeat sequences within the plastomes. This software detects regions where certain sequences are repeated directly adjacent to one another, which can be important for understanding structural variation. The tool was used with default parameters, making it straightforward to identify and characterize these tandem repeats. For the detection of microsatellite loci, MISA-web47 (https://webblast.ipk-gatersleben.de/misa/) was utilized. MISA-web is a powerful tool for identifying simple sequence repeats (SSRs) or microsatellites, which are short, repetitive DNA sequences that can be highly variable and are often used in genetic studies. The parameters were set to identify a minimum of 10 mononucleotide repeats, 5 dinucleotide repeats, 4 trinucleotide repeats, and 3 repeats for tetra-, penta-, and hexanucleotide motifs. Additionally, microsatellite loci were considered compound loci if the distance between two microsatellites was less than 100 bp, allowing for the detection of more complex repeat structures.
Codon usage bias analysis
We used Geneious Prime 2021, a comprehensive bioinformatics software, to extract protein-coding gene sequences from the seven plastomes of A. kurzii and M. scandens. The extracted sequences were required to be at least 300 bp and have a total number of bases divisible by three to ensure proper codon alignment. Additionally, duplicate sequences were filtered out to maintain data quality and accuracy.
To analyze codon usage bias for both A. kurzii and M. scandens, we utilized CodonW1.4.4 software (https://codonw.sourceforge.net/). CodonW is a specialized tool for analyzing codon usage patterns and bias across different organisms. In addition, we calculated codon usage bias parameters, including T3s (thymine at the third position), C3s (cytosine at the third position), A3s (adenine at the third position), G3s (cytosine at the third position), CAI (Codon Adaptation Index), CBI (Codon Bias Index), Fop (frequency of optimal codons), ENC (effective number of codons), and GC3s (GC content at the third position of codons), for the protein-coding genes of the seven sampled plastomes.
We used the R package to generate ENC-plot and PR2-plot graphs to facilitate a better comparison of codon usage bias differences between A. kurzii and M. scandens. To assess the influence of mutation pressure and selection pressure on codon usage bias, we compared the expected ENC values with the observed ENC values48. Additionally, the PR2-plot was used to represent the codon usage bias of each gene based on the nucleotide composition of the third position of the codons (A, T, C, G), providing insights into the balance between mutation and selection. Finally, we analyzed the optimal codons of the two species, which were selected based on the following criteria: difference in RSCU between the two species (ΔRSCU) > 0.08 and relative synonymous codon usage (RSCU) > 1, indicating a preference for certain codons in the species’ genetic code.
Selection pressure analysis
The Geneious Prime 2021 was used to extract the shared protein-coding sequences (80 CDSs) from the seven plastomes of A. kurzii and M. scandens. This process involved aligning the sequences to generate a sequence matrix, ensuring that only homologous sequences were included for comparative analysis. Subsequently, the synonymous substitution rate (dN) and nonsynonymous substitution rate (dS) were calculated using DnaSP v5.0 software, and the dN/dS ratio was computed using Excel. The dN/dS ratio, a key indicator of selective pressure (where a ratio greater than 1 suggests positive selection, and a ratio less than 1 suggests purifying selection). Finally, the analyzed protein-coding genes were functionally classified based on the results of the dN/dS ratio calculations, and multiple comparisons visualization was generated using the “ggplot2” package in R v.4.0.1. The “ggplot2” is a powerful data visualization package in R that allows for the creation of complex and informative plots, facilitating the comparison and interpretation of the data across different functional gene groups.
Phylogenomic analyses
The plastome sequences of 24 samples (as listed in Table S1) were used for constructing a phylogenetic tree, employing both Maximum Likelihood (ML) and Bayesian Inference (BI) methods, with B. nivea and C. sativa as outgroups. Initially, duplicate protein-coding genes from all plastomes were filtered out to ensure data accuracy. Multiple sequence alignment was performed using MAFFT v7.48749, a widely used tool for aligning multiple sequences with high accuracy, which resulted in the creation of an information matrix necessary for subsequent analyses.. The ML phylogenetic tree was constructed using IQ-TREE 2.0.550 which implemented the TVM + F + I + G4 model, a specific model of nucleotide substitution. To ensure the robustness of the tree, 1000 UFBoot Bootstrap resampling and 1000 SH tests were conducted. The best-fit model and parameters for the phylogenetic analysis were determined using the online tool CIPRES51,52, in conjunction with ModelTest v3.753. This process identified the optimal GTR + I + G model, which was then used to generate the final Bayesian phylogenetic tree through MCMC simulations conducted for 1 million generations, with sampling every 1000 generations.
Divergence time estimation was carried out using BEAST v1.8.053,54, a software package designed for Bayesian analysis of molecular sequences, which allows for the estimation of divergence times and evolutionary rates. An uncorrelated relaxed clock model was applied as the molecular clock model, and the Yule Model was used as the tree prior model. The phylogenetic tree was calibrated using fossil age information from previous studies3, with the root node for Broussonetia set at 33.9 million years ago (Mya) and the root node for Ficus calibrated at 56 Mya3. The resulting tree, which included divergence times, was visualized and enhanced using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/), a tool for viewing and annotating phylogenetic trees.
For genetic diversity analysis, the 80 shared protein-coding sequences (CDSs) were extracted from the plastomes of each species. The nucleotide diversity (pi) values were calculated using DnaSP v5.0 software. Subsequently, the pi values were classified into three regions based on their values in descending order: > 0.02 (high), 0.01–0.02 (medium), and < 0.01 (low). The sequences from these regions were concatenated to form matrices, which were then imported into BEAUTi for further parameter settings. The partition parameter was set to include a joint tree. The uncorrelated relaxed clock was utilized as the molecular clock model, and the Yule Model was employed as the tree prior model. The substitution model for each partition was GTR + I + G, and a normal distribution was chosen for the prior distribution. A total of 500,000,000 Markov Chain Monte Carlo (MCMC) iterations were conducted, with samples taken every 1,000 iterations. Convergence of the chains was evaluated using Tracer v1.7.155, where parameters with effective sample sizes (ESS) greater than 200 were considered reliable. Additionally, Tree Annotator v1.10.4 was used to discard the initial 10% of the samples. The resulting tree file, which included divergence times, was visualized and enhanced using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Data availability
The plastomes of Allaeanthus kurzii and Malaisia scandens generated in this study are available in the NCBI GenBank repository (details in Table S1). Specifically, for Allaeanthus kurzii, the experiment numbers are MSN1 and MSN2, with GeneBank accession numbers PP584575 and PP577929, respectively. For Malaisia scandens, the experiment numbers are MSC1, MSC2, and MSC3, with GeneBank accession numbers PP584597, PP577928, and PP728628, respectively.
References
Gardner, E. M. et al. Repeated parallel losses of inflexed stamens in Moraceae: Phylogenomics and generic revision of the tribe Moreae and the reinstatement of the tribe Olmedieae (Moraceae). Taxon 70, 946–988 (2021).
Chung, K. F. et al. Molecular recircumscription of Broussonetia (Moraceae) and the identity and taxonomic status of B. kaempferi var. australis. Bot. Stud. 58, 11 (2017).
Zhang, Q., Onstein, R. E., Little, S. A. & Sauquet, H. Estimating divergence times and ancestral breeding systems in Ficus and Moraceae. Ann. Bot. 123, 191–204 (2018).
Tangkanakul, P., Trakoontivakorn, G., Auttaviboonkul, P., Niyomvit, B. & Wongkrajang, K. Antioxidant activity of northern and northeastern thai foods containing indigenous vegetables. Kasetsart J. (Nat. Sci.) 40, 47–58 (2006).
Thwaites, G. H. K. Descriptions of some new genera and species of ceylon plants: Allaeanthus. Hooker’s J. Bot. Kew Gard. 6, 302–303 (1854).
Zerega, N. Flora malesiana (Moraceae-Ficus). by C. C. Berg and E J. H. corner. Brittonia 58, 194–195 (2006).
Corner, E. J. H. The classification of Moraceae. Gard Bull Singapore 19, 187–252 (1962).
Kuo, W. H. et al. Plastome phylogenomics of Allaeanthus, Broussonetia and Malaisia (Dorstenieae, Moraceae) and the origin of B. × kazinoki. J. Plant Res. 135, 203–220 (2022).
Kim, M. & Zavada, M. S. Pollen morphology of Broussonetia (Moraceae). Grana 32, 327–329 (1993).
Clement, W. L. & Weiblen, G. D. Morphological evolution in the mulberry family (Moraceae). Syst. Bot. 34, 530–552 (2009).
Gardner, E. M. et al. Phylogenomics of Brosimum (Moraceae) and allied genera, including a revised subgeneric system. Taxon 70, 778–792 (2021).
Zerega, N. J. C. & Gardner, E. M. Delimitation of the new tribe Parartocarpeae (Moraceae) is supported by a 333-gene phylogeny and resolves tribal level Moraceae taxonomy. Phytotaxa 388, 253 (2019).
Wolfe, K. H., Li, W. H. & Sharp, P. M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. PNAS 84, 9054–9058 (1987).
Wu, C. S., Chen, C. I. & Chaw, S. M. Plastid phylogenomics and plastome evolution in the morning glory family (Convolvulaceae). Front. Plant Sci. 13, 1061174 (2022).
Tonti-Filippini, J., Nevill, P. G., Dixon, K. & Small, I. What can we do with 1000 plastid genomes?. Plant J. 90, 808–818 (2017).
Ruhfel, B. R., Gitzendanner, M. A., Soltis, P. S., Soltis, D. E. & Burleigh, J. G. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 14, 23 (2014).
Zhu, A. D., Guo, W. H., Gupta, S., Fan, W. S. & Mower, J. P. Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 209, 1747–1756 (2016).
Moran, E. V., Willis, J. & Clark, J. S. Genetic evidence for hybridization in red oaks (Quercus sect. Lobatae, Fagaceae). Am. J. Bot. 99, 92–100 (2012).
Gitzendanner, M. A., Soltis, P. S., Yi, T. S., Li, D. Z. & Soltis, D. E. Plastome phylogenetics: 30 years of inferences into plant evolution. In Plastid Genome Evolution (eds Chaw, S. M. & Jansen, R. K.) 293–313 (Academic Press, 2018).
Nadine, T. & Bock, R. The translational apparatus of plastids and its role in plant development. Mol. Plant 7, 1105–1120 (2014).
Barrett, C. F. et al. Investigating the path of plastid genome degradation in an early-transitional clade of heterotrophic orchids, and implications for heterotrophic angiosperms. Mol. Biol. Evol. 31, 3095–3112 (2014).
Kim, K. J. & Lee, H. L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261 (2004).
Millen, R. S. et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658 (2001).
Cummings, H. S. & Hershey, J. W. Translation initiation factor IF1 is essential for cell viability in Escherichia coli. J. Bacteriol. 176, 198–205 (1994).
Liu, M. et al. Complete genome sequence of a Chinese isolate of pepper vein yellows virus and evolutionary analysis based on the CP, MP and RdRp coding regions. Arch. Virol. 161, 677–683 (2016).
Hubert, F. et al. Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus. Syst. Biodivers. 12, 405–423 (2014).
Matsuoka, Y., Yamazaki, Y., Ogihara, Y. & Tsunewaki, K. Whole chloroplast genome comparison of rice, maize, and wheat: implications for chloroplast gene diversification and phylogeny of cereals. Mol. Biol. Evol. 19, 2084–2091 (2002).
Hebert, P. D. N., Ratnasingham, S. & deWaard, J. R. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Royal Soc. Lond. B 270(Suppl 1), S96-99 (2003).
Zerega, N. J. C., Clement, W. L., Datwyler, S. L. & Weiblen, G. D. Biogeography and divergence times in the mulberry family (Moraceae). Mol. Phylogenet. Evol. 37, 402–416 (2005).
Wang, L. & Roossinck, M. J. Comparative analysis of expressed sequences reveals a conserved pattern of optimal codon usage in plants. Plant Mol. Biol. 61, 699–710 (2006).
Mohasses, F. C., Solouki, M., Ghareyazie, B., Fahmideh, L. & Mohsenpour, M. Correlation between gene expression levels under drought stress and synonymous codon usage in rice plant by in-silico study. PLoS ONE 15, e0237334 (2020).
Parvathy, S. T., Udayasuriyan, V. & Bhadana, V. Codon usage bias. Mol. Biol. Rep. 49, 539–565 (2022).
Plotkin, J. B. & Kudla, G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 (2011).
Huang, Y. et al. Comparative analysis of Diospyros (Ebenaceae) plastomes: insights into genomic features, mutational hotspots, and adaptive evolution. Ecol. Evol. 13, e10301 (2023).
Morton, B. R. The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 56, 616–629 (2003).
Pahlich, E. & Gerlitz, C. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemistry 19, 11–13 (1980).
Qin, H. et al. Threatened species list of China’s higher plants. Biodiv. Sci. 25, 696–744 (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Jin, J. J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241 (2020).
Lohse, M., Drechsel, O. & Bock, R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274 (2007).
Amiryousefi, A., Hyvönen, J. & Poczai, P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031 (2018).
Brudno, M. et al. LAGAN and multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13, 721–731 (2003).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Kurtz, S. et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642 (2001).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Fuglsang, A. Impact of bias discrepancy and amino acid usage on estimates of the effective number of codons used in a gene, and a test for selection on codon usage. Gene 410, 82–88 (2008).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Miller, M. A. et al. A RESTful API for access to phylogenetic tools via the CIPRES science gateway. Evol. Bioinf. 11, 43–48 (2015).
Ronquist, F. et al. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Drummond, A. J., Nicholls, G. K., Rodrigo, A. G. & Solomon, W. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161, 1307–1320 (2002).
Drummond, A. J. & Rambaut, A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Funding
This work was funded by the Agricultural Resources and Environmental Protection Project- Survey of Agricultural Wild Plant Resources (Grant no. 13220104), Forestry and Grassland Ecological Protection and Restoration Fund (National Park Subsidy) Project -Integrated Survey and Monitoring of Resources in Hainan Tropical Rainforest National Park (Grant No. HDZB-2023-071) and the Research Funds for the Natural Science Foundation of Zhejiang Province (Grant No. LY21C030002).
Author information
Authors and Affiliations
Contributions
Y. H. Zhang, Y. Q. Chen., and X.J. Jin conceived and designed the study; L.N. Zhou and L.X. Yuan performed the experiments and data analysis; L. X. Yuan, P. Li, J. R. Lei, Z. Z. Chen, and Z. H. Zhang contributed to material collection; L.N. Zhou wrote the first manuscript; Y. H. Zhang, L.X. Yuan, X.J. Jin, P. Li, and B. L. Wei edited the manuscript. All authors have approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Since neither the deciduous flower mulberry nor the cow tendon vine are endangered species of higher plants in China, nor were they collected from protected areas, no authorization was required. Our plant collection and experimental procedures adhered to relevant institutional, national, and international guidelines and legislation. The collected samples are housed at the Herbarium of Wenzhou University (WZU), with voucher numbers: Pan Li 010350-Pan Li 010354. All samples were identified by the second author (Lang-Xing Yuan) and the third author (Pan Li).
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, LN., Yuan, LX., Li, P. et al. Comparative analyses of plastomes in Allaeanthus and Malaisia: structure, evolution, and phylogeny. Sci Rep 14, 22686 (2024). https://doi.org/10.1038/s41598-024-73941-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-73941-4
