
Abstract
In conservation strategies, getting precise and repeatable information on the species’ diet and health without relying on invasive or laborious methods is challenging. Here, we developed an efficient and non-invasive workflow for the sequencing and analysis of four taxonomic markers from fecal DNA to characterize the gut microbiota, parasites, and plants and lichens composing the winter diet of caribou (Rangifer tarandus), Canada’s most iconic endangered species. Sequencing of the 18S rRNA gene of eukaryotes from seven locations in Manitoba and Saskatchewan, Canada, allowed for the detection of five genera of parasites in caribou feces (including Nematodirella and Parelaphostrongylus) with variable frequency of occurrence depending on sampling location and sex. Our workflow also revealed a rich winter plant and lichen diet in caribou, with respectively 29 and 18 genera identified across all samples through plant and fungal ITS2 sequencing. Relationships between the gut microbiota and both the diet and parasite richness were also identified. Of note, the Central Saskatchewan sampling location was characterized by a clearly distinct gut microbiota which could be linked to an epiphytic lichen-rich diet. Overall, our results showed the potential of this multi-marker DNA metabarcoding workflow as an efficient tool to provide insights into the species biology and ecology.
Similar content being viewed by others

Introduction
With ongoing global climate change, many wildlife species are vulnerable or at risk of extinction1,2. Among them, caribou (Rangifer tarandus), an iconic wildlife species, is declining across its circumpolar distribution3. Boreal caribou, in particular, ranges across the entire boreal ecozone and is currently listed as threatened under Canada’s Species At Risk Act, with only fifteen out of fifty-one local populations being self-sustaining4.
The main reasons identified for the decline of boreal caribou are disturbances and modifications to its habitat due to human activities5,6, in addition to direct and indirect effects of climate change7. Modifications to caribou habitat can favor the presence of other ungulates species and predators8,9 and affect the composition of the ground vegetation. The Boreal caribou diet is composed of plants and lichens, the latter being the main source of energy during winter10. As a result, caribou forage in peatland, open lichen woodland and old growth coniferous forests11,12, three ecosystems that are experiencing modifications in response to climate change13,14. Suitable nutritional gain is crucial for caribou survival, particularly during the winter season, when foraging might be difficult, and for pregnant females that rely on lichens, a highly digestible source of energy15. As an example, intake of Salix by Svalbard caribou was found to be positively correlated to autumn body mass condition, which is in turn linked to winter survival16. Deepening our knowledge of the caribou diet could help us better protect its habitat, but so far this has proved difficult: collar camera studies are complex and involve low number of individuals17 and previous attempts to characterize the diet based on environmental DNA from fecal samples mostly failed to recover lichens18,19.
Like all ruminants, caribou rely on microbes for efficient and successful digestion and their entire digestive tract has evolved to exploit microbial fermentation. Therefore, potential changes in diet could impact its gut microbial community structure and functions16,20. The composition of the gut microbiota is also linked to health and metabolism in numerous mammal species16,21,22,23 as well as range size in ungulates24. Moreover, climate change and modification of habitat could impact caribou-parasite interactions25,26,27. A higher presence and abundance of parasites could be linked to a decrease in fitness28,29,30 and be particularly deleterious for declining populations. Finally, low quality nutrition can lead to greater susceptibility to parasite infection and create a vicious circle where animals with poor body condition are more likely to be infected, which in turn affects their fitness31. In this context, simultaneous knowledge about the animal’s diet, gut microbiota, and parasites would be beneficial.
For an endangered species with a very large geographic range like caribou, a standardized and high-throughput workflow to study these various aspects of the biology and ecology of the species should be developed to limit over-interpretation of differences among studies that could arise from methodological biases. Metabarcoding of multiple taxonomic markers, including those covering major plant and lichen diet components, is a promising approach to achieve this goal. Metabarcoding of DNA isolated from feces is an effective and non-invasive way to describe the gut microbiota, as exemplified by previous studies on caribou16,20,32,33, and can also be applied to the detection of parasites that inhabit mammals’ digestive tract34,35,36. It is also commonly being used to describe the diet of various organisms37,38,39,40. Moreover, there is good potential for synergy between this approach and existing genetic surveys, with both cost and time savings accrued by extracting more information from material already collected during ongoing fecal collection campaigns, like through the EcoGenomics research program, and creating the possibility of linking diet, parasite and microbiota data to specific individuals through time. However so far, while the caribou plant diet was successfully characterized by metabarcoding of fecal DNA18,19,41, previous studies either did not address the lichen diet16,41 or failed to do it because of methodological issues19 or lack of sequences in databases18, leading to an important gap that this study is proposing to fill.
While metabarcoding is increasingly being used to describe diet, gut microbiota and parasites, the focus of each study is generally limited to one or two of these aspects and the method is applied to small sample sizes. Moreover, variations in the methodology make the merging of metabarcoding datasets from different studies challenging42. In this study, we propose a standardized workflow, from sampling to data analysis, to describe caribou diet, gut microbiota and parasites from caribou feces using the ITS2 regions of fungi and plants, the 16S rRNA gene of prokaryotes and the 18S rRNA gene of eukaryotes, as taxonomic markers. The workflow was developed using fecal pellets from 96 individuals among seven locations across central Canada. Fecal pellets were collected during the winter season to ensure optimal conditions for the preservation of DNA and advance knowledge related to the winter diet, which is critical for caribou but remains poorly documented10.
Materials and methods
Sample selection
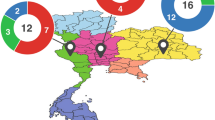
Fecal pellets from 96 individuals sampled during the winter (January to March) in seven locations in Manitoba and Saskatchewan, Canada, were selected from an existing extensive collection held by the EcoGenomics Team and used to generate population demographic parameters43. Samples from four to eight males and females from each sampling location were included in the analysis (Fig. 1, Supplementary Table S1). Individuals were identified by genotyping based on variable microsatellite loci amplified from fecal DNA, as previously described44,45,46,47, and caribou-specific primers Zfx/Zfy were used for sex identification45. Samples were shipped on dry ice and stored at -80 °C, during 2.5 to 6.5 years, prior to DNA isolation for metabarcoding analyses.

Caribou fecal pellets sampling locations in Saskatchewan and Manitoba, Canada. Map was generated using QGIS version 3.22.4-Białowieża, https://qgis.org/. Ecozone shapefiles were obtained from the Government of Canada https://sis.agr.gc.ca/cansis/nsdb/ecostrat/gis_data.html.
Selection of taxonomic markers and primers
Appropriate taxonomic markers and corresponding primers were selected to ensure the detection of groups of organisms of interest from caribou fecal DNA. Broad coverage 16S rRNA gene primers targeting the V4-V5 regions48 were selected to characterize the prokaryotic microbiota (i.e., bacteria and archaea). Parasites colonizing the caribou digestive tract are members of various taxonomic groups, including the Apicomplexa, Archamoebae, Platyhelminthes, Nematoda, and Arthropoda49. To simultaneously detect all these organisms, broad coverage primers targeting the 18S rRNA gene of eukaryotes50 were selected. Moreover, these primers have the potential to provide information on the caribou’s winter diet, which is composed of lichens ̶ a symbiotic relationship that minimally includes a fungus and a phototroph (typically an alga)51 ̶ and plants10. They could also detect anaerobic fungi of the Neocallimastigomycota phylum, which colonizes the gastrointestinal tract of ruminants and play an important role in their digestion52. More specific markers commonly used to identify lichens and plants were also selected for the characterization of the diet. As the identification of lichens is based on the taxonomy of the core fungal symbiont53, which can be achieved through the sequencing of the nuclear ribosomal internal transcribed spacer (ITS)54, primers targeting the fungal ITS2 region were selected55,56. The Neocallimastigomycota phylum would also be detected by these primers. For plants, no single widely used marker gene is currently accepted, but sequencing of the plant ITS2 region has been shown suitable for a large variety of plants, allow for an appropriate taxonomic identification at the genus level57, and is supported by well-developed gene databases58. Previously published primers targeting the plant ITS2 were therefore included in the selection59. A list of all primers used in this study is provided in Supplementary Table S2.
DNA isolation, library preparation, and sequencing
For each individual sample, three frozen fecal pellets were ground in liquid nitrogen using a mortar and pestle. DNA isolation was performed on 100 mg of ground homogenized samples using the DNeasy PowerSoil kit (QIAGEN, Netherlands) according to the manufacturer’s protocol (May 2017) for QIACube instruments (QIAGEN). The Qubit 3.0 fluorometer (Thermo Fisher Scientific, MA, USA) was used for the quantitation of DNA using the dsDNA BR (broad range) assay kit (Life Technologies, CA, USA). DNA extracts were diluted to 5 ng/µL with 10 mM Tris pH 8.0 solution using the QIAgility automated system (QIAGEN).
Library preparation for Illumina sequencing was conducted following the manufacturer’s protocol (16S Metagenomic Sequencing Library Preparation, Part 15044223 Rev. B) with some modifications. The 5’ end of the primers (Supplementary Table S2) was flanked with one or the other of the following Illumina overhang adapter sequences: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG (forward overhang) and GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG (reverse overhang). PCR reactions were set up by mixing 25.0 µL of 2X HotStarTaq Plus Master Mix (QIAGEN, Germany), 0.5 µL of each 10 µM HPLC-purified (or desalted) primer (Invitrogen, USA), 19 µL of RNase-free water (QIAGEN) and 5.0 µL of DNA diluted at 5 ng/µL, for a total volume of 50 µL. Thermal cycling conditions for the 16S rRNA gene were as follows: initial denaturation at 95 °C for 5 min; 35 cycles (40 cycles for the 18S, fungal ITS2 and plant ITS2) at 94 °C for 45 s, 50 °C (47 °C for 18S, 50 °C for fungal ITS2 and 49 °C for plant ITS2) for 45 s, 72 °C for 1 min; and a final elongation step at 72 °C for 10 min. PCR products were visualized on GelRed-stained 1% agarose gel, using the ChemiGenius Bioimaging System (Syngene, Cambridge, UK). Target fragment sizes were ~ 479 bp for the 16S rRNA gene, ~ 484 bp for the 18S rRNA gene, ~ 347–487 bp for the fungal ITS2 region and ~ 230–378 bp for the plant ITS2 region.
PCR products were purified using AMPure XP reagent (Agencourt, Beckman Coulter Life Science, CA, USA) according to Illumina’s protocol, with gentle pipetting as the preferred option for mixing. Combinatorial dual-index barcodes were added by amplifying 5 µL of the purified PCR product with 25 µL of 2X KAPA HIFI HotStart Ready mix (Roche, South Africa), 5 µL of each Nextera XT index primer (Nextera XT Index kits v2, Illumina Inc., CA, USA) and 10 µL of RNase-free water, for a total volume of 50 µL. Thermal cycling conditions were as follows: 3 min at 95 °C; 10 cycles of 30 s at 95 °C, 30 s at 55 °C, 30 s at 72 °C; and a final elongation step of 5 min at 72 °C. The resulting dual-indexed libraries were purified with the AMPure XP reagent following the procedure described above, quantified using the SYNERGY Mx system (BioTek Instruments, VT, USA) and pooled at equimolar ratio. To compensate for low base diversity in libraries, 15% of PhiX Control V3 (Illumina) was spiked in the pool. Sequencing was performed using a Miseq v3 600 cycle kit on an Illumina MiSeq system at the Next-Generation Sequencing Platform, Genomics Centre, CHU de Québec-Université Laval Research Centre, Québec, Canada.
Pipeline development and database optimization
The pipeline Q2Pipe was developed to ensure a user-friendly and standardized treatment of amplicon sequencing data with QIIME260. Briefly, QIIME2 and its dependencies were loaded to a Singularity image (https://zenodo.org/records/4667718), which allows users to work in a batch environment by installing Singularity, downloading the Singularity image and cloning the Q2Pipe Github repository (https://github.com/NRCan/Q2Pipe). Users can also build the Singularity image by using the image recipe provided in the Q2Pipe repository. The newly built pipeline allows for the selection and modification of all options available in QIIME2 in a single text file which can easily be shared among users and published, while also offering the option to run secondary external programs like FUNGuild61, directly from the pipeline. For compatibility purposes, Q2Pipe can also be used with the QIIME2 Anaconda installation method if the user installs the external dependencies in the Anaconda virtual environment.
As several errors have been detected in the genus name of Eukaryota within the SILVA SSU database 138–9962, a custom python script was used to screen and correct taxonomical disparities between the name at level 6 (i.e. genus) and the binomial name at level 7 (i.e. species). This correction affected 56% of the Eukaryota genera (not applied to Bacteria and Archaea). Furthermore, a low number of entries for lichens were observed in the UNITE database63, which is intended for the molecular identification of fungi based on sequencing of the ITS region. To improve the taxonomic assignment for this group, UNITE was enriched with sequences from the OLICH database64 as well as from 20 lichen specimens collected in Newfoundland, Canada, and formally identified based on anatomical/morphological features65. Protocols for DNA isolation, amplification, sequencing and bioinformatic treatment of sequences from lichen samples was as described above for fecal samples. Only ITS2 region sequences corresponding to the genus identified by anatomical/morphological features and representing more than 10% of sequences from a sample were retained. This resulted in 22 lichen sequences from 19 species that have been added to the customized database: Alectoria sarmentosa, Cetraria aculeata, Cladonia amaurocraea, Cladonia arbuscula, Cladonia gracilis var gracilis, Cladonia maxima, Cladonia rangiferina (three sequences from two specimens), Cladonia squamosa, Cladonia stellaris, Cladonia uncialis, Lasalia papulosa, Lobaria pulmonaria, Lobaria quercizans, Platismatia norvegica, Sphaerophorus globosus, Stereocaulon tomentosum, Tuckermannopsis americana and Usnea filipendula. Lichen sequence data and metadata have been deposited in the Genbank database of the National Center for Biotechnology Information (NCBI) (accession numbers OQ792092 to OQ792112).
Bioinformatics analyses on sequences from caribou fecal samples
Bioinformatics analyses for each sequence dataset obtained from caribou feces were performed using QIIME2, version 2020.8 within Q2Pipe. Option files for each gene analyzed can be found as Supplementary Material 1. Although parameters were slightly different for each taxonomic marker, the overall procedure was similar. Briefly, demultiplexed, per-sample, gzipped fastq files were imported and sequencing reads were truncated at their 5’ and 3’ ends based on (1) the length of the primer (16S and 18S) or specific primer sequence (ITS2) and (2) per base sequence quality score. The QIIME2 plugin DADA266 was selected as the denoising method, which allowed for filtering, dereplication, merging paired-end reads and chimera identification. This resulted in inferences of amplicon sequence variants (ASVs). The least abundant ASVs were filtered out if their frequency was less than 0.05% of the mean ASV frequency. Naive Bayes trained classifier were generated from the corrected and amended SILVA SSU database 138–9962, the amended UNITE v8.3 dynamic fungal database63, and the PLANiTS v2020.3 database58. Taxonomic assignment of 16S and 18S rRNA gene ASVs (SILVA database), fungal ITS2 ASVs (UNITE database) and plant ITS2 ASVs (PLANiTS) was then performed using the QIIME2 plugin feature-classifier classify-sklearn, which is a machine-learning-based classification method that requires trained classifiers. For the 16S rRNA gene, ASVs assigned to Eukaryota, mitochondria, chloroplast and unassigned ASVs at the kingdom level were filtered out, while ASVs from Bacteria, Mammalia and unassigned ASVs at the kingdom level were filtered out for the 18S rRNA gene. For plant and fungal ITS2 regions, taxonomic filtering was performed to keep only either Streptophyta or Fungi. ASV tables of the 18S rRNA gene and fungal ITS2 region were further analyzed using FUNGuild61 to infer the ecological guilds of fungi based on their taxonomy. Metabarcoding sequence data and metadata have been deposited in the Sequence Read Archive of the NCBI (BioProject PRJNA904780, accession numbers SRR23356982 to SRR23357367).
Characterization of the diet and detection of potential parasites
Potential components of the diet were filtered from non-rarefied ASV tables among plants (plant ITS2 and 18S datasets), lichen-forming fungi (fungal ITS2 and 18S datasets) and lichen-forming algae (18S dataset). Lichen-forming fungal ASVs were identified based on FUNGuild trophic mode assignment, while ASVs from lichen-forming algae were identified based on the list published by Sanders & Masumoto67. The following steps were carried out on filtered datasets containing only ASVs belonging to these potential components of the diet. To increase the reliability of our data and discard potential contaminants, ASVs were considered as part of the diet when accounting for more than 1% of the sequences in a sample68. Then, relative abundance data was converted to presence/absence, as numerous factors may lead to bias in sequence representation of food items in DNA isolated from fecal samples69. The frequency of occurrence (i.e. the number of samples that contain a given food item, referred to here as FOO), as proposed by Ando et al.70, was used as a semi-quantitative approach to underline diet differences between locations. Plant and lichen ASVs were classified according to their morphology, hereby called guilds for plants (herbaceous, non-vascular or lignified) or growth forms for lichens (crustose, epiphyte foliose, epiphyte fruticose, foliose or fruticose). The number of ASVs assigned to plants and lichens as well as to each plant guild and lichen growth form were calculated and used as a proxy for the richness of these various components of the diet.
To identify parasites, ASVs belonging to the Apicomplexa, Archamoebae, Platyhelminthe, Nematoda and Arthropoda were extracted from the non-rarefied 18S rRNA ASV table; ASVs from these groups were then listed and parasites were identified based on the list of caribou parasites from Tryland and Kutz49. ASVs assigned to the Entamoeba genus, a common parasite in mammals, were also included in the parasite list even if, to the best of our knowledge, it has never been reported as a caribou parasite. Moreover, ASVs assigned to the Blastocystis genus, a ubiquitous anaerobic protist which develops in the gastrointestinal tract of a wide variety of hosts such as mammals (including some ungulates), birds and reptiles71,72 were also included, although it belongs to the Stramenopila lineage73 and was not identified through our initial taxa selection. As errors in the taxonomic assignment of Eukaryotes was previously observed with the SILVA database, each taxonomic assignment was verified by conducting a BLAST analysis of ASV sequences against the NCBI Genbank database. When the BLAST results were not concordant with the taxonomic assignment obtained at the genus level by our bioinformatic pipeline, the sequences were discarded from the list. As a result, all sequences assigned to the Cryptosporidium genus were discarded as they matched to uncultured eukaryote clones, the first Cryptosporidium hit in NCBI sharing only around 91% similarity with sequences from our dataset. Overall, five genera of potential parasites were detected and included in further analyses. As for the diet, parasite detection was described by the frequency of occurrence and the number of ASVs assigned to each genus as a proxy for the richness.
Statistical analyses
All statistical analyses were performed in R (4.2.2 Patched) using 0.05 as a significance threshold value.
For alpha and beta diversity analyses, datasets were normalized by rarefaction in QIIME2 to control for uneven sequencing across samples74 (see Q2Pipe Option files in Supplementary Material 1 for details). Optimal library size for each taxonomic marker was selected considering both the rarefaction curves and the number of reads of the smallest libraries. Sampling depth was set at 18,430 sequences for the 16S rRNA gene, resulting in 1,769,280 sequences clustered into 7,192 ASVs observed among 96 samples (100%); 2,750 sequences for the 18S rRNA gene dataset, resulting in 264,000 sequences clustered into 5,174 ASVs observed among 96 samples (100%); 3,700 sequences for the plant ITS2 region, resulting in 351,500 sequences clustered into 1,585 ASVs observed among 95 samples (99%); and 7,100 sequences for the fungal ITS2 region, resulting in 674,500 sequences clustered into 7,326 ASVs observed among 95 samples (99%). The ASV richness (Chao1 index) and diversity (Shannon index) for all markers were estimated with the “vegan” package. To evaluate differences in these indices as well as in richness (i.e., number of ASVs) of different functional groups (parasites, plant diet and lichen diet) among locations and between sex, Kruskal-Wallis tests (kruskal.test function) were performed, followed by Dunn post hoc test (dunn.test function) when significant. The same approach was used to assess differences in the relative abundance of various taxonomic groups (i.e. eukaryotic, fungal and bacterial phyla, plant families, and the twenty most abundant genera of each dataset) among locations and between sex. Correlations between the relative abundance of the twenty most abundant bacterial genera (i.e., bacterial genera with the highest mean relative abundance across all samples) and the richness of various components of the diet (i.e., number of ASVs assigned to lichens belonging to Crustose, Epiphytic Foliose, Epiphytic Fruticose, Foliose, and Fruticose growth forms; number of ASVs assigned to Herbaceous, Non-vascular, or Lignified plants) and of parasites (total number of ASVs assigned to parasites; number of ASVs assigned to each parasite genus detected in more than 25% of the samples) were also tested using Spearman’s Rank correlation (cor.mtest function). Correlations between the relative abundance of the Neocallimastigomycota phylum and the same diet and parasite richness estimates were also assessed using Spearman’s Rank correlation.
The effects of sampling locations and sex on beta diversity for each marker gene were tested by permutational multivariate analysis of variance (PERMANOVA) using the “adonis” function of the “vegan” package based on Bray-Curtis dissimilarity matrices obtained from the “vegdist” function. When significant, differences between sampling locations were tested by pairwise PERMANOVA using the “pairwise.adonis” function. Non-metric multidimensional scaling (NMDS) plots were generated for each taxonomic marker using the “metaMDS” function, based on Bray-Curtis dissimilarity to visualize differences among locations and between sex. The “envfit” function of the “vegan” package was used to test whether the parasite, plant and lichen richness were correlated to the bacterial NMDS space. Only significant correlations were mapped on the NMDS plots.
Results
Taxonomic composition and diversity detected by each taxonomic marker
18S rRNA gene
As expected, sequencing of the 18S rRNA gene allowed for the detection of all three Eukaryote kingdoms. A total of 44 phyla were detected across the seven locations. The Phragmoplastophyta phylum had the highest relative abundance (21.3%), followed by the Archamoebae (17.4%), Chlorophyta (11.2%) and Nematoda (10.9%) phyla (Fig. 2a). Significant differences in the relative abundance of several phyla among locations and between sex were detected (Fig. 2a, Supplementary Table S3A). For example, the relative abundance of Chlorophyta (green algae) was lowest for Central Saskatchewan and Wabowden and highest for The Bog. The relative abundance of Nematoda was higher in males, while the relative abundance of Archamoebae was higher in females.

Relative abundance of eukaryotic phyla (a) and prokaryotic genera (b) detected in caribou fecal DNA from seven locations based on the sequencing of the 18S and 16S rRNA genes, respectively. Only the 20 phyla (a) or genera (b) having the highest mean relative abundance across all samples are shown. Bog = The Bog (MB), Char = Charron Lake (MB), CSK = Central Saskatchewan (SK), Flin = Flin Flon (SK), Nao = Naosap-Reed (MB), Ron = La Ronge (SK) and Wab = Wabowden (MB). Black and pink stars indicate significant differences in relative abundance of a taxa between locations and sex, respectively (see Supplementary Tables S3A and S6B for supporting statistical information).
Eukaryotic richness (Chao1 index) and diversity (Shannon index) were generally higher in La Ronge when compared to the other locations (Supplementary Table S3B). The composition of the eukaryotic community also differed among locations (Fig. 3a) and the variance in beta diversity was mainly explained by this factor (R2 = 0.205, p = 0.001) and, to a lesser extent, by sex (R2 = 0.034, p = 0.001). All Adonis pairwise comparisons between locations were significant, except for Flin Flon, which was only different from Central Saskatchewan, and Naosap-Reed and Wabowden, which were not significantly different (Supplementary Table S3C).

NMDS plots showing the variation in beta diversity between sex and locations (based on Bray-Curtis dissimilarities), according to (a) 18S rRNA gene, (b) plant ITS2 region, (c) fungal ITS2 region and (d) 16S rRNA gene sequences from caribou fecal DNA. Arrows in panel d) indicate correlations between the NMDS space and richness of various components of the diet (i.e., number of ASVs assigned to lichens belonging to the Crustose, Epiphytic Foliose, Epiphytic Fruticose, and Foliose growth forms; number of ASVs assigned to Herbaceous, Non-vascular, or Lignified plants) and parasites (total number of ASVs assigned to a parasite genus; number of ASVs assigned to the Blastocystis and Entamoeba genera). Only correlations that were identified as significant (p < 0.05) by the envfit function are shown (see Supplementary Table S6E for envfit results of all variables tested).
The 18S rRNA gene is not commonly used to detect and describe plant and fungal communities, as more specific markers are available and provide better taxonomic resolution for these groups. Correspondingly here, only 55% of the Phragmoplasmophyta sequences of the 18S rRNA gene dataset were assigned at the genus level. Of the 24 plant genera detected, only 10 (Equisetum, Juncus, Polytrichum, Populus, Rhynchospora, Scheuchzeria, Sphagnum, Ticodendron, Triglochin, Typha, Urtica) passed the 1% threshold used to identify components of the diet. Considering lichen forming algae, 11 genera were detected by the sequencing of the 18S rRNA gene and eight of them (Asterochloris, Chlamydomonas, Coccomyxa, Elliptochloris, Leptosira, Symbiochloris, Trebouxia, Trentepohlia) passed the 1% threshold. Only one ASV assigned to a lichen-forming fungus was detected. Considering the low number of plant genera detected in the 18S rRNA gene dataset (when compared to the plant ITS2 marker) and the poor performance of this marker for the detection and identification of lichens, only the plant and fungal ITS2 markers were considered in further diet analyses.
Plant ITS2 region
A total of 45 plant families were detected based on the sequencing of the ITS2 region, with Ericaceae as the most abundant (39.28%), followed by Cyperaceae (16.84%), Sphagnaceae (7.24%), Hylocomiaceae (5.13%) and Betulaceae (4.74%) (Supplementary Fig. S1). Another 8.57% of the sequences were not assigned at the family level. The majority of the twenty most abundant plant families showed differences in their relative abundances among locations (Supplementary Table S4A). As an example, the relative abundance of Ericaceae was much higher at The Bog and Central Saskatchewan than in other locations.
No difference in plant richness was found among locations (Supplementary Table S4B). However, the Shannon diversity index was significantly higher in Naosap-Reed than in Central Saskatchewan. As for the 18S rRNA gene, beta diversity based on the plant ITS2 sequencing was significantly influenced by the sampling location (Fig. 3b, R2 = 0.237, p = 0.001). All Adonis pairwise comparisons between locations were significant, except for Naosap-Reed and Flin Flon which were similar (Supplementary Table S4C). No difference in plant alpha diversity was detected between sex.
Fungal ITS2 region
A total of nine fungal phyla were detected, with Ascomycota as the most abundant (65.24%), followed by Basidiomycota (16.91%) and Neocallimastigomycota (7.57%) (Supplementary Fig. S2). Unidentified fungi represented 9% of the total number of ASVs. Some differences in relative abundances among locations were detected (Supplementary Table S5A), such as the higher relative abundance of Ascomycota in Central Saskatchewan when compared to La Ronge and Wabowden.
Fungal richness (Chao1) was higher in Central Saskatchewan than in Naosap-Reed, The Bog and Wabowden (Supplementary Table S5B) and higher for females than for males. Shannon diversity index was not significantly different among locations but was higher for females than for males (Supplementary Table S5B). The fungal beta diversity was primarily driven by location (R2 = 0.198, p = 0.001), with Central Saskatchewan samples clustering separately from the others (Fig. 3c). Adonis pairwise comparisons showed that all locations were significantly different from each other, except for Flin Flon which was similar to Wabowden, Naosap-Reed and The Bog (Supplementary Table S5C), a trend that can be visualized in Fig. 3c.
16S rRNA gene
A total of 39 bacterial phyla were detected, with Firmicutes as the most abundant (72.38%), followed by Bacteroidota (23.59%), Proteobacteria (0.94%), Verrucomicrobiota (0.94%) and Cyanobacteria (0.58%) (Supplementary Fig. S3). Some differences in the relative abundance among locations were detected, but mostly in phyla having a low relative abundance (< 1%) (Supplementary Table S6A).
The richness (Chao1) and the Shannon diversity index were slightly lower in La Ronge and Central Saskatchewan than in other locations (Supplementary Table S6C). As for other markers, the variance of beta diversity of the 16S rRNA gene was explained by location (R²=0.218, p = 0.001) while sex was nearly significant (R²=0.012, p = 0.076) (Fig. 3d). All Adonis pairwise comparisons between locations were significant, except for Flin Flon which was similar to The Bog and Naosap-Reed (Supplementary Table S6D).
Characterization of the caribou parasites, diet and gut microbiota based on multi-marker metabarcoding data
Parasites
Five genera that host potential caribou parasites were detected in the non-rarefied 18S rRNA gene dataset: Eimeria (Apicomplexa), Nematodirella and Parelaphostrongylus (Nematoda), Entamoeba (Archamoebae), and Blastocystis (Stramenopila lineage). Entamoeba was the most frequently detected and was found in all locations, followed by Parelaphostrongylus and Blastocystis (Fig. 4). Eimeria and Nematodirella were only detected in two and three locations, respectively (Fig. 4). Differences in the number of ASVs assigned to parasitic genera among locations were also detected: Naosap-Reed had more Entomoeba ASVs and, overall, more ASVs assigned to parasitic genera than other locations, Charron Lake had more Nematodirellla ASVs, while Flin Flon and Naosap-Reed had more Blastocystis ASVs (Fig. 5a, Supplementary Table S3D). Nematodes (Parelaphostrongylus and Nematodirella) were more frequently found in males than in females (Fig. 4) and the number of ASVs assigned to these genera was also higher in males than in females (Supplementary Tables S3D). However, no difference was found between males and females for the total parasite richness.

Heatmap of the frequency of occurrence of five parasite genera detected in caribou fecal DNA based on the sequencing of the 18S rRNA gene, by sex and location. Bog = The Bog (MB), Char = Charron Lake (MB), CSK = Central Saskatchewan (SK), Flin = Flin Flon (SK), Nao = Naosap-Reed (MB), Ron = La Ronge (SK) and Wab = Wabowden (MB). F = Female, M = Male.

Number of ASVs of (a) parasites, (b) plants and (c) lichens detected in caribou fecal DNA based on sequencing of the 18S rRNA gene as well as plant and fungal ITS2 regions, respectively, by location. Bog = The Bog (MB), Char = Charron Lake (MB), CSK = Central Saskatchewan (SK), Flin = Flin Flon (SK), Nao = Naosap-Reed (MB), Ron = La Ronge (SK) and Wab = Wabowden (MB).
Diet
Of the 1,585 plant ASVs detected by the sequencing of the plant ITS2 marker, 148 ASVs assigned to 29 genera passed the 1% threshold and were kept for diet analysis. For the lichens, a total of 603 ASVs were detected in the fungal ITS2 dataset, of which 382 ASVs, assigned to 18 lichen genera, passed the 1% threshold and were kept for diet analysis. The FOO was calculated for each plant and lichen genus and separate heatmaps were generated to assess how the plant (Supplementary Figure S4) and lichen (Supplementary Figure S4) diets differed among locations. A third heatmap combining FOO data from the 15 most frequently detected plant genera and the 15 most frequently detected lichen genera was also generated to evaluate the usefulness of an integrated characterization of the diet (Fig. 6).

Heatmap of the frequency of occurrence of plant and lichen genera composing the caribou diet based on the sequencing of the plant and fungal ITS2 regions from fecal DNA, by location and sex. Only the 15 most frequently detected plants and lichens genera detected across all samples were included in the analysis. Bog = The Bog (MB), Char = Charron Lake (MB), CSK = Central Saskatchewan (SK), Flin = Flin Flon (SK), Nao = Naosap-Reed (MB), Ron = La Ronge (SK) and Wab = Wabowden (MB). F = Female, M = Male.
Analysis of the FOO of plant genera identified as part of the diet resulted in three plant groups (Supplementary Figure S4). The first group consisted of the most frequently detected plant genera: Andromeda, Kalmia, Carex, Pleurozium and Sphagnum, which had high FOOs in almost all, if not all, locations. The second group was composed of Rhododendron, Betula and Salix and could be defined as a “secondary diet” for which the FOOs were lower but similar for all locations. The third group was composed of plant genera that were not frequently detected in caribou fecal DNA (Arctostaphylos, Calliergon, Chamaedaphne, Comarum, Eriophorum, Gaylussacia, Juncus, Juniperus, Larix, Menyanthes, Picea, Pinus, Polytrichum, Populus, Potentilla, Ptilidum, Rubus, Sarmentypnum, Sarracenia, Triglochin, Vaccinium) or that had high FOO in a single or few locations, such as Populus and Polytrichum at La Ronge or Sarracenia and Potentilla at The Bog. Based on this analysis of the FOO of plant genera, the La Ronge location stood out as the most distinct in terms of plant diet (Supplementary Figure S4). Analysis of the richness of various plant guilds (using the number of ASVs assigned to taxa from each guild as a proxy) (Fig. 5b, Supplementary Table S4D) further showed a high number of non-vascular plant ASVs and a low number of lignified plant ASVs at La Ronge than in other locations, while the opposite trend was observed at The Bog. In contrast, high numbers of both non-vascular plant and lignified plant ASVs were observed in Central Saskatchewan. No difference in plant diet was found between sex (Supplementary Figure S4 and Table S4D).
The analysis of the FOO of lichens also resulted in three groups (Supplementary Figure S5). The first group consisted of two genera that were frequently detected in all locations: Cladonia and Bryoria. The second group was composed of Usnea, Evernia, Hypogymnia and Parmelia, epiphytic lichens found in all locations, but at overall higher FOOs in Central Saskatchewan and Wabowden (females only) than in other locations. The third group was composed of lichen genera detected at low FOOs in most locations (Buellia, Flavopunctelia, Lecanora, Micarea, Parmeliopsis, Phylliscum, Plectenia, Raesaeneniana, Ramalina, Sarea, Stereaucolon, Violella), except for Raesaeneniana and Stereocaulon in La Ronge and Charron Lake (males only). Based on the FOO of lichen genera, the CSK and La Ronge locations appeared as the most distinct (Supplementary Figure S5). The total number of ASVs assigned to lichens was lower at The Bog and higher in Central Saskatchewan and La Ronge. The number of ASVs for crustose, epiphyte-foliose and epiphyte-fruticose lichens was also higher in Central Saskatchewan whereas the number of ASVs for fruticose lichens was lower (Fig. 5c, Supplementary Table S5D). Sex had no impact on the number of lichen ASVs (Supplementary Figure S5).
The combined analysis of the plant and lichen diets (Fig. 6) showed consistent trends with the analyses performed separately, although the different groups of plant and lichen genera described above were not as clearly defined. The La Ronge and Central Saskatchewan locations were again identified as the most distinct (Fig. 6). Sex had a lower impact on the diet than location, most male and female samples from the same location clustering together, except for the Wabowden location (Fig. 6).
Gut microbiota
Neocallimastigomycota fungi, which are an important component of the gut microbiota of ruminants, were detected both by 18S and fungal ITS2 sequencing, although relative abundances were lower in the 18S dataset. A strong correlation was however detected between the 18S and ITS relative abundance values (Supplementary Figure S6), indicating that both markers could be used for the detection of this phyla. The relative abundance of Neocallimastigomycota was influenced by the location, with samples from Central Saskatchewan having the lowest relative abundance in both the 18S and ITS2 datasets. The number of Neocallimastigomycota ASVs was also impacted by location based on the 18S dataset, with higher values in Naosap-Reed and The Bog than in Central Saskatchewan, while in the ITS2 dataset the location effect was not significant (Supplementary Table S3E). Sex had no effect on the relative abundance or the number of Neocallimastigomycota ASVs.
Several common bacteria of the gut microbiota of ruminants were detected using 16S rRNA gene sequencing. Among the most abundant at the genus level were the Ruminococcaceae UCG-005 (19.95%) and UCG-010 (9.26%), Bacteroides (11.8%), and Christensenellaceae R-7 group (5.26%) (Fig. 2b). The Central Saskatchewan and La Ronge locations were the most distinct, with significant differences in relative abundance detected for eight out of the 20 top bacterial genera (Supplementary Table S6B). Of note, the relative abundance of Prevotella was much lower in Central Saskatchewan compared to other locations, but higher for Christensenellaceae R7 (Fig. 2b, Supplementary Table S6B). Analysis of correlations between the NMDS space of the prokaryotic beta diversity and diet richness indicators showed that the distinct community observed in Central Saskatchewan was linked to the dissimilar diet observed in this location (i.e., higher diversity of epiphytic lichens) (Fig. 3d, Supplementary Table S6E). The richness of Entamoeba and Blastocystis as well as the total parasite richness were also linked to the structure of the gut microbial community (Fig. 3d, Supplementary Table S6E).
ASV richness of some plant guilds and lichen growth forms was significantly correlated to the relative abundance of the Neocallimastigomycota phylum and of some of the most abundant bacterial genera (Fig. 7). Of note, the diversity of epiphytic lichens was positively correlated to the relative abundance of the Ruminococcaceae UCG 005 and Christensenellaceae R7 group but negatively correlated to the relative abundance of Prevotella and UCG 010 (Firmicutes, Oscillospirales class). Significant correlations were also detected between the richness of parasites and the relative abundance of some bacterial genera (Fig. 7).

Correlations between the relative abundance of the twenty most abundant bacterial genera (i.e., bacterial genera with the highest mean relative abundance across all samples) and of the Neocallimastigomycota phylum (X-axis) and the richness of various components of the diet (i.e., number of ASVs assigned to lichens belonging to Crustose, Epiphytic Foliose, Epiphytic Fruticose, Foliose, and Fruticose growth forms; number of ASVs assigned to Herbaceous, Non-vascular, or Lignified plants) and of parasites (total number of ASVs assigned to parasites; number of ASVs assigned to the Blastocystis, Entamoeba and Parelaphostrongylus genera) (Y-axis) from caribou fecal DNA. Red and blue colors are used to indicate the correlation coefficient only if the correlation is significant.
Discussion
This study showed that multi-marker metabarcoding of DNA isolated from non-invasively collected fecal pellets has the potential to generate new insight into the biology and ecology of at-risk species such as caribou. Our optimized workflow is based on four taxonomic markers with a broad coverage: the 16S rRNA gene for prokaryotes, 18S rRNA gene for eukaryotes and ITS2 regions of plants and fungi. This resulted in the detection of genera that include caribou parasite species and in a broad characterization of the caribou winter diet (including both plants and lichens) and of the gut microbiota (bacteria and anaerobic fungi). Performance of the four taxonomic markers and the potential of applying our multi-marker metabarcoding workflow to address research questions related to the caribou biology and ecology are discussed.
Performance of selected taxonomic markers
Sequencing of the 18S rRNA gene for the identification of various functional groups (i.e., parasites, plants, lichens) from diverse phyla present in ruminant feces had, to our knowledge, only been attempted once in a very controlled environment75 prior to this study. For most animal species, the diet is restricted to a few groups of organisms and specific marker genes are generally used70. Here, by targeting the 18S rRNA gene, plants and lichens as well as parasites were simultaneously detected in an herbivorous species. Five genera of potential parasites belonging to the phyla Apicomplexa, Archamoebae, Nematoda and to the Stramenopila lineage were identified in caribou feces using this approach: Eimeria, Nematodirella, Parelaphostrongylus, Entamoeba, and Blastocystis. Species-level identification of parasites was not possible based on the sequencing of the V4 region of the 18S rRNA gene but would have been of great interest for some of these genera. For example, the Parelaphostrongylus genus includes at least three species identified as caribou parasites (P. odocoilei, P. tenuis, and P. andersoni) but their frequency of occurrence and impact on caribou health is quite different: P. andersoni, a muscle worm infecting caribou and other Cervidae in Canada, is common in boreal caribou, whereas P. tenuis, the meningeal worm, is rare but lethal26. Other complementary approaches, such as the sequencing of the ITS2 region using primers specifically designed to target nematodes34 or the use of qPCR detection assays allowing to differentiate species, as proposed for other nematodes (see Reslova76 et al. as an example), could be considered in this case. Several common parasitic nematodes such as Varestrongylus eleguniensis77, Ostertagia gruehneri29 or Platyhelminte species such as Moniezia benedeni78 were expected, given the relatively high prevalence reported in above-mentioned studies, but went undetected in our fecal samples. This could be linked to the time of sampling, as parasitic infections are known to be seasonal in many species including caribou78. Here, sample collection was exclusively done during the winter to ensure integrity of the fecal DNA in the context of caribou genetic studies.
Although this seasonal bias could be a limitation for the detection of some parasites by our workflow, the fact that sequencing of the 18S rRNA gene allowed to detect some known genera of caribou parasites, but also genera that were not previously reported, indicate that this approach could contribute to the detection of emerging parasites and be useful in the context of a biosurveillance strategy. Of note, the genus Entamoeba, which include parasite species that could be pathogenic, such as Entamoeba hystolyticain human79, or with a level of pathogenicity which is still unresolved, such as Entamoeba bovisin ruminants80, was observed at high frequencies in caribou feces from all locations. While no study to date had reported Entamoebainfections in caribou, new hosts are frequently identified81. Another new potential parasite detected in caribou feces is Blastocystis, found in six of the seven locations. Reindeer (Rangifer tarandus) in northeastern China as well as other wild ungulates, including white-tailed deer (Odocoileus virginianus) in the USA, have also been identified as hosts of this ubiquitous protist82,83,84,85. Further studies will be needed to identify the Entamoeba species colonizing caribou and assess the impact of both Entamoeba and Blastocystis, if any, on caribou health.
While useful to provide information about potential parasites, sequencing of the 18S rRNA gene was found to be less informative than more specific taxonomic markers for the characterization of the caribou winter diet. Among the fungal ASVs detected, only one was associated with a lichen-forming fungus. Lichens (and probably more so the fungal symbiont) are highly digestible for caribou15 and it is therefore possible that the fungal lichen DNA remaining in feces was not abundant enough (when compared to DNA from other eukaryotes) to be detected at the sequencing depth applied in this study. Alternatively, primers bias, a common issue in metabarcoding studies86, may have favored the amplification of the 18S rRNA gene of other groups of eukaryotes. Nevertheless, several genera of lichen-forming algae were detected by our workflow, but the information provided was less precise, as the same algal species can form various lichens, and the lichen taxonomy is therefore based on the taxonomy of the fungal symbiont53. In contrast, sequencing of the ITS2 region provided great insight into the caribou lichen diet based on fecal DNA, which had not been previously described for caribou using a metabarcoding approach due to methodology failures18,19. Surprisingly, despite the use of a conservative threshold of 1% to include ASVs into the diet, 18 lichen genera were detected across all samples, which is higher than the 13 lichen genera reported by Webber et al.10 across 30 different studies based on the histological analysis of caribou feces or rumen content, indicating that DNA-based approaches are likely more sensitive. Enrichment of the fungal ITS2 database used for taxonomic assignment of sequences is most likely one of the reasons why we had better success in using metabarcoding to detect and identify lichen in caribou fecal DNA. This improvement opens the door to future studies on the caribou lichen diet.
Description of the plant diet based on the 18S rRNA gene was also not as informative as the plant ITS2 region: a much lower proportion of the plant sequences detected in the 18S dataset were assigned to a genus than in the ITS2 dataset (55% vs. 91%) and the number of genera identified as part as the diet was also lower (10 vs. 29). Overall, while the reasons behind these discrepancies among taxonomic markers still need to be identified, these results indicate that ITS2 sequencing is a more suitable approach for plant and lichen identification from fecal DNA, allowing for the detection of a rich winter diet in boreal caribou. These findings further highlight the importance of using stable and consistent methods to assess the diet and infer ecological links based on metabarcoding data, as the use of various markers can lead to different findings.
Metabarcoding of the 16S rRNA gene is a widely used approach for the characterization of the gut prokaryotic microbiota and did not require much optimization. The fungal microbiota, and more specifically anaerobic fungi of the Neocallimastigomycota phylum, which plays an important role in the digestion of ruminants52, is however less frequently studied. Here, two of the taxonomic markers analyzed provided sequences that could be assigned to the Neocallimastigomycota phylum. A good correlation was found in the relative abundances measured by the two markers, indicating that both could be used to provide information about this taxonomic group.
Insights into caribou biology and ecology based on multi-marker metabarcoding of fecal DNA
Despite the relatively limited number of samples analyzed in this study, the results provided some insights on the potential of using our metabarcoding workflow to address research questions related to caribou biology and ecology. Considering the diet, plant genera growing as groundcover (Carex, Pleurozium and Sphagnum) or in the understory forest layer (Andromeda and Kalmia) were the most frequently detected. The presence of mosses and Ericaceae in the diet have already been described using metabarcoding of fecal DNA in migratory caribou18 and in several other studies assessing the caribou diet using histological methods, as reviewed by Webber et al.10. However, Thompson et al.17 observed, using collar cameras, that the proportion of mosses and Ericaceae in the boreal caribou’s winter diet was much lower than that of lichens, an information that can not be obtained from metabarcoding data. In terms of lichens, as expected, Cladonia and Bryoria were the most frequently detected as they are commonly reported as important food items for caribou10,17.
Differences among the seven sampling locations were clearly revealed by the analysis of both the plant and lichen diets. The La Ronge location could mostly be distinguished from other locations by its lower frequency of ericaceous shrubs (Kalmia and Andromeda) but higher diversity of non-vascular plants composing the diet. Two lichen genera, Stereocaulon and Raesaeneniana, were also more frequently detected at La Ronge than in other locations. This shift in the diet may be associated with the transition between boreal plain and boreal shield forest at this location87, indicating that boreal caribou adapts its plant diet to what is found in its environment10. The Central Saskatchewan location was also distinct in terms of lichen diet, with epiphytic lichens detected more frequently and at a higher richness than in other locations, whereas soil fruticose lichens had lower frequencies and richness. As the diversity of the fruticose growth form is mostly driven by Cladonia in our dataset, it can be assumed that caribou from Central Saskatchewan had limited access to this main food source during the winter17. Two hypotheses could be put forward to explain why caribou from this location would forage more than others on epiphytic lichens and plants composing the midstory vegetation layer. First, caribou dig less on terrestrial lichens as the snow thickness and density increase during the winter88 or in the presence of ice crust that prevent caribou from grazing on the ground, as recently demonstrated in Peary caribou89. Second, the region is under intensive forest management, an activity known to affect the caribou’s winter diet17,90. Although further studies will be needed to identify drivers of caribou diet in the La Ronge and Central Saskatchewan locations, our findings clearly demonstrate the importance of considering lichens when studying the winter diet of caribou, something that our workflow allows to successfully do using a DNA-based approach.
Interestingly, while being different from other locations in terms of lichen diet, the Central Saskatchewan location was also characterized by its clearly distinct microbiota. Noteworthy, lower relative abundance and diversity of anaerobic fungi of the Neocallimastigomycota phylum were detected in fecal DNA from this location. Neocallimastigomycota fungi have been linked to fibrous plant digestibility and lignin degradation91, and diet was shown to be a key factor influencing these fungal populations52. Their low abundance within the gut microbial community of caribou in Central Saskatchewan is interesting as these samples had the most diverse lignified plant diet and a higher relative abundance of Ericaceae. On the one hand, this could be linked to a digestive bias, as less digestible food items have more chances to be detected in feces70, while on the other hand, the lack of Neocallimastigomycota fungi in the gut microbiota can lead to a decrease in fiber digestion potential, as demonstrated in sheep92, which could have further improved the detection of these less digestible food items in feces from the Central Saskatchewan location.
As previously described in reindeer32,93,94, the gut prokaryotic microbiota of caribou was mainly composed of Firmicutes and Bacteroidota in our study, and several of the most abundant taxa detected were also found as dominant in other ruminants worldwide95, supporting that a core microbiota exists in ruminants. Interesting correlations between the gut prokaryotic microbiota and the diet were detected, confirming trends observed in other ruminants20,95,96. Beta diversity of the prokaryotic community was correlated with the richness of the lichen diet and, to a lesser extent, of the plant diet. Of note, the distinct cluster formed by Central Saskatchewan samples in the ordination correlated with a higher richness in epiphytic lichen, further demonstrating that the specific diet found at this location is influencing the caribou microbiota. Richness of the lichen diet was also better correlated than the richness of the plant diet to the relative abundance of several taxa known for their functional role in the metabolism and fermentation process in ruminants52,97,98, with both positive (e.g., Christensenellaceae R7 Group) and negative (e.g., Prevotella, Neocallimastigomycota) correlations to the richness of epiphytic lichens detected. These important bacterial and fungal taxa of the caribou digestive tract could potentially be proposed as indicators of the quality of the diet, and eventually of the habitat, but more research is needed on this front.
While strong differences among sampling locations were detected for most of the variables analyzed in this study, differences between sex were more limited. One interesting observation made on that end was that nematodes belonging to the genera Nematodirella and Parelaphostrongylus were more frequently detected and had a higher richness in males than in females. The trend was not tested independently for each location, given the limited number of individuals per location, but the pattern seemed to be present in at least three locations (Central Saskatchewan, Charron Lake and Flin Flon) and would deserve further investigation. In mammals, it is recognized that males are typically more parasitized than females due to their behavior and biology, a fact that could be linked to male-biased mortality99. Here, the gut microbial community was also linked to the diversity of parasites, indicating that parasites could also play a role on caribou health through its impact on the gut microbiota. As indicated by studies performed on livestock (including ruminants) in the context of helminth infections, reviewed by William et al.100, while both diet and parasite infections can alter the gut microbiota, diet-induced modifications can also affect the host susceptibility to parasites. The workflow proposed here, which allows simultaneous characterization of the diet, parasites, and gut microbiota from fecal DNA, could greatly contribute to the study of the complex interactions between these various factors, and eventually lead to the identification of robust individual and population-level health indicators.
Conclusion
Overall, through the optimization of the various steps of the workflow, including the development of a tool allowing to simplify and standardize the bioinformatic analyses of sequences and the enrichment of the fungal ITS2 database with sequences from lichenous fungi, this work has allowed for the implementation of an efficient, non-invasive, and standardized multi-marker metabarcoding workflow for the characterization of caribou fecal DNA. The application of this workflow to samples from seven locations in Manitoba and Saskatchewan highlighted its capacity to reveal similarities and differences between locations, especially for the winter diet, as well as to establish associations between datasets, such as those detected between the lichen diet and the gut microbiota. These results showed that the development of diet and health indicators derived from the metabarcoding of caribou fecal DNA, in parallel to host genetic data, is a promising approach to gather relevant information in the context of population dynamics studies, including niche partitioning and effects of natural and anthropogenic disturbances and habitat loss assessment, therefore providing critical information to adequately protect boreal caribou and its habitat.
Data availability
Lichen sequence data and metadata have been deposited in the Genbank database of the National Center for Biotechnology Information (NCBI) (accession numbers OQ792092 to OQ792112). Metabarcoding sequence data and metadata have been deposited in the Sequence Read Archive of the NCBI (BioProject PRJNA904780, accession numbers SRR23356982 to SRR23357367).
References
Arneth, A. et al. Post-2020 biodiversity targets need to embrace climate change. Proc. Natl. Acad. Sci. 117, 30882–30891 (2020).
Urban, M. C. Accelerating extinction risk from climate change. Science. 348, 571–573 (2015).
Vors, L. S. & Boyce, M. S. Global declines of caribou and reindeer. Glob Change Biol. 15, 2626–2633 (2009).
Environment and Climate Change Canada. Amended recovery strategy for the Woodland Caribou (Rangifer tarandus caribou), Boreal population, in Canada [Proposed]. Species at Risk Act Recovery Strategy Series, xiii + pp. 143 (Environment and Climate Change Canada, Ottawa, 2019). https://wildlife-species.canada.ca/species-risk-registry/virtual_sara/files/plans/Rs-CaribouBorealeAmdMod-v01-2020Dec-Eng.pdf.
Festa-Bianchet, M., Ray, J. C., Boutin, S., Côté, S. D. & Gunn, A. Conservation of caribou (Rangifer tarandus) in Canada: an uncertain future. Can. J. Zool. 89, 419–434 (2011).
Stewart, F. E. C. et al. Boreal caribou can coexist with natural but not industrial disturbances. J. Wildl. Manag. 84, 1435–1444 (2020).
Neilson, E. W. et al. The direct and habitat-mediated influence of climate on the biogeography of boreal caribou in Canada. Clim. Change Ecol. 100052https://doi.org/10.1016/j.ecochg.2022.100052 (2022).
Serrouya, R., McLellan, B. N., Boutin, S., Seip, D. R. & Nielsen, S. E. Developing a population target for an overabundant ungulate for ecosystem restoration. J. Appl. Ecol. 48, 935–942 (2011).
Wittmer, H. U., Mclellan, B. N., Serrouya, R. & Apps, C. D. Changes in landscape composition influence the decline of a threatened woodland caribou population. J. Anim. Ecol. 76, 568–579 (2007).
Webber, Q. M. R., Ferraro, K. M. & Hendrix, J. G. Vander Wal, E. What do caribou eat? A review of the literature on caribou diet. Can. J. Zool. 100, 197–207 (2022).
Leblond, M., Dussault, C. & St-Laurent, M. H. Development and validation of an expert-based habitat suitability model to support boreal caribou conservation. Biol. Conserv. 177, 100–108 (2014).
Rettie, W. J. & Messier, F. Hierarchical habitat selection by woodland caribou: its relationship to limiting factors. Ecography. 23, 466–478 (2008).
Payette, S. & Delwaide, A. Tamm review: the North-American Lichen Woodland. Ecol. Manag. 417, 167–183 (2018).
Price, D. T. et al. Anticipating the consequences of climate change for Canada’s boreal forest ecosystems. Environ. Rev. 21, 322–365 (2013).
Parker, K. L., Barboza, P. S. & Stephenson, T. R. Protein conservation in female caribou (Rangifer tarandus): effects of decreasing diet quality during winter. J. Mammal. 86, 610–622 (2005).
Kamenova, S. et al. Arctic greening drives changes in the diet and gut microbiome of a resident herbivore with consequences for fitness. bioRxiv. 2024.02.27.582426 https://doi.org/10.1101/2024.02.27.582426 (2024).
Thompson, I. D. et al. Factors influencing the seasonal diet selection by woodland caribou (Rangifer tarandus tarandus) in boreal forests in Ontario. Can. J. Zool. 93, 87–98 (2015).
Brodeur, A., Leblond, M., Brodeur, V., Taillon, J. & Côté, S. D. investigating potential for competition between migratory caribou and introduced muskoxen. J. Wildl. Manag. 87, e22366 (2023).
Mitchell, G. et al. DNA metabarcoding of faecal pellets reveals high consumption of yew (Taxus spp.) by caribou (Rangifer tarandus) in a lichen-poor environment. FACETS. 7, 701–717 (2022).
Salgado-Flores, A. et al. Rumen and cecum microbiomes in reindeer (Rangifer tarandus tarandus) are changed in response to a lichen diet and may affect enteric methane emissions. PLoS One 11, e0155213 (2016).
Desselberger, U. The mammalian intestinal microbiome: composition, interaction with the immune system, significance for vaccine efficacy, and potential for disease therapy. Pathogens. 7, 57 (2018).
Kinross, J. M., Darzi, A. W. & Nicholson, J. K. Gut microbiome-host interactions in health and disease. Genome Med. 3, 14 (2011).
Moeller, A. H. & Sanders, J. G. Roles of the gut microbiota in the adaptive evolution of mammalian species. Philos. Trans. R Soc. B Biol. Sci. 375, 20190597 (2020).
Wolf, J. F. et al. Gut microbiome composition predicts summer core range size in two divergent ungulates. FEMS Microbiol. Ecol. 97, fiab048 (2021).
Laaksonen, S. et al. Climate change promotes the emergence of serious disease outbreaks of filarioid nematodes. EcoHealth. 7, 7–13 (2010).
Kutz, S. J. et al. Parasites in ungulates of Arctic North America and Greenland: a view of contemporary diversity, ecology, and impact in a world under change. Adv. Parasitol. 79, 99–252. https://doi.org/10.1016/B978-0-12-398457-9.00002-0 (2012)
Morales-Castilla, I. et al. Forecasting parasite sharing under climate change. Philos. Trans. R Soc. B Biol. Sci. 376, 20200360 (2021).
Albon, S. D. et al. The role of parasites in the dynamics of a reindeer population. Proc. R Soc. Lond. B Biol. Sci. 269, 1625–1632 (2002).
Dickinson, E. R., Orsel, K., Cuyler, C. & Kutz, S. J. Life history matters: Differential effects of abomasal parasites on caribou fitness. Int. J. Parasitol. 53, 221–231 (2023).
Irvine, R. J., Corbishley, H., Pilkington, J. G. & Albon, S. D. Low-level parasitic worm burdens may reduce body condition in free-ranging red deer (Cervus elaphus). Parasitology. 133, 465–475 (2006).
Beldomenico, P. M. & Begon, M. Disease spread, susceptibility and infection intensity: vicious circles? Trends Ecol. Evol. 25, 21–27 (2010).
Ilina, L. A. et al. The comparative analysis of the ruminal bacterial population in reindeer (Rangifer tarandus L.) from the Russian Arctic zone: regional and seasonal effects. Animals. 11, 911 (2021).
Pope, P. B. et al. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS One 7, e38571 (2012).
Avramenko, R. W. et al. Exploring the gastrointestinal nemabiome: deep amplicon sequencing to quantify the species composition of parasitic nematode communities. PLOS ONE. 10, e0143559 (2015).
Gasser, R. B., Bott, N. J., Chilton, N. B., Hunt, P. & Beveridge, I. Toward practical, DNA-based diagnostic methods for parasitic nematodes of livestock — bionomic and biotechnological implications. Biotechnol. Adv. 26, 325–334 (2008).
Vlčková, K. et al. Diversity of Entamoeba spp. in African great apes and humans: an insight from Illumina MiSeq high-throughput sequencing. Int. J. Parasitol. 48, 519–530 (2018).
Erickson, D. L. et al. Reconstructing a herbivore’s diet using a novel rbcL DNA mini-barcode for plants. AoB Plants. 9, plx015 (2017).
Li, J., Zhan, S., Liu, X., Lin, Q., Jiang, J., & Li, X. Divergence of fecal microbiota and their associations with host phylogeny in Cervinae. Front. Microbiol. 9, (2018).
McShea, W. J. et al. Metabarcoding reveals diet diversity in an ungulate community in Thailand. Biotropica. 51, 923–937 (2019).
Srivathsan, A., Ang, A., Vogler, A. P. & Meier, R. Fecal metagenomics for the simultaneous assessment of diet, parasites, and population genetics of an understudied primate. Front. Zool. 13, 17 (2016).
Christopherson, V., Tremblay, J. P., Gagné, P. N., Bérubé, J. & St-Laurent, M. H. Meeting caribou in the alpine: do moose compete with caribou for food? Glob Ecol. Conserv. 20, e00733 (2019).
O’Rourke, D. R., Bokulich, N. A., Jusino, M. A., MacManes, M. D. & Foster J. T. A total crapshoot? Evaluating bioinformatic decisions in animal diet metabarcoding analyses. Ecol. Evol. 10, 9721–9739 (2020).
Hettinga, P. N. et al. Estimating size and trend of the North Interlake Woodland caribou population using fecal-DNA and capture–recapture models. J. Wildl. Manag. 76, 1153–1164 (2012).
Ball, M. C., Finnegan, L., Manseau, M. & Wilson, P. Integrating multiple analytical approaches to spatially delineate and characterize genetic population structure: an application to boreal caribou (Rangifer tarandus caribou) in central Canada. Conserv. Genet. 11, 2131–2143 (2010).
Ball, M. C. et al. Characterization of target nuclear DNA from faeces reduces technical issues associated with the assumptions of low-quality and quantity template. Conserv. Genet. 8, 577–586 (2007).
McFarlane, S., Manseau, M. & Wilson, P. J. Spatial familial networks to infer demographic structure of wild populations. Ecol. Evol. 11, 4507–4519 (2021).
Priadka, P. et al. Partitioning drivers of spatial genetic variation for a continuously distributed population of boreal caribou: implications for management unit delineation. Ecol. Evol. 9, 141–153 (2019).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Tryland, M. & Kutz, S. J. (eds.). Reindeer and Caribou: Health and Disease, 1st edn. (CRC Press, 2018). https://doi.org/10.1201/9780429489617.
Stoeck, T. et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31 (2010).
Spribille, T., Resl, P., Stanton, D. E. & Tagirdzhanova, G. Evolutionary biology of lichen symbioses. New. Phytol. 234, 1566–1582 (2022).
Gruninger, R. J. et al. Anaerobic fungi (phylum neocallimastigomycota): advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol. Ecol. 90, 1–17 (2014).
Lücking, R., Hodkinson, B. P. & Leavitt, S. D. The 2016 classification of lichenized fungi in the Ascomycota and Basidiomycota – approaching one thousand genera. Bryologist. 119, 361–416 (2017).
Kelly, L. J. et al. DNA barcoding of lichenized fungi demonstrates high identification success in a floristic context. New. Phytol. 191, 288–300 (2011).
White, T. J., Bruns, T., Lee, S. & Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications 315–322. (Academic Press, 1990). https://doi.org/10.1016/B978-0-12-372180-8.50042-1
Menkis, A. et al. Occurrence and impact of the root-rot biocontrol agent Phlebiopsis gigantea on soil fungal communities in Picea abies forests of northern Europe. FEMS Microbiol. Ecol. 81, 438–445 (2012).
Hollingsworth, P., Graham, S. & Little, D. Choosing and using a plant DNA barcode. PloS One. 6, e19254 (2011).
Banchi, E. et al. PLANiTS: a curated sequence reference dataset for plant ITS DNA metabarcoding. Database 2020, baz155 (2020).
Chen, S. et al. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 5, e8613 (2010).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Nguyen, N. H. et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Abarenkov, K. et al. The UNITE database for molecular identification of fungi – recent updates and future perspectives. New. Phytol. 186, 281–285 (2010).
Marthinsen, G., Rui, S. & Timdal, E. OOLICH: A reference library of DNA barcodes for Nordic lichens. Biodivers. Data J. 7, e36252. https://doi.org/10.3897/BDJ.7.e36252 (2019).
Hill, D. J. Keys to lichens of North America: revised and expanded. Lichenologist. 48, 255–255 (2016).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 13, 581–583 (2016).
Sanders, W. B. & Masumoto, H. Lichen algae: the photosynthetic partners in lichen symbioses. Lichenologist. 53, 347–393 (2021).
Ando, H. et al. Evaluation of plant contamination in metabarcoding diet analysis of a herbivore. Sci. Rep. 8, 15563 (2018).
Pompanon, F. et al. Who is eating what: diet assessment using next generation sequencing. Mol. Ecol. 21, 1931–1950 (2012).
Ando, H. et al. Methodological trends and perspectives of animal dietary studies by noninvasive fecal DNA metabarcoding. Environ. DNA. 2, 391–406 (2020).
Jiménez, P., Muñoz, M. & Ramírez, J. D. An update on the distribution of Blastocystis subtypes in the Americas. Heliyon 8(12), e12592. https://doi.org/10.1016/j.heliyon.2022.e12592 (2022).
Anamalai, S. et al. Blastocystis in captivated and free-ranging wild animals worldwide: a revie. Trop. Biomed. 39, 338–372 (2022).
Grattepanche, J. D. et al. Microbial diversity in the eukaryotic SAR Clade: illuminating the darkness between morphology and molecular data. BioEssays. 40, 1700198 (2018).
Schloss Patrick, D. Rarefaction is currently the best approach to control for uneven sequencing effort in amplicon sequence analyses. mSphere 9, e00354-23 (2024).
Kamenova, S. et al. DNA metabarcoding diet analysis in ruminants is quantitative and integrates feeding over several weeks. bioRxiv. 2024.02.01.577814 https://doi.org/10.1101/2024.02.01.577814 (2024).
Reslova, N. et al. The identification and semi-quantitative assessment of gastrointestinal nematodes in faecal samples using multiplex real-time PCR assays. Parasit. Vectors. 14, 391 (2021).
Verocai, G. G. et al. The biogeography of the caribou lungworm, Varestrongylus eleguneniensis (Nematoda: Protostrongylidae) across northern North America. Int. J. Parasitol. Parasites Wildl. 11, 93–102 (2020).
Turgeon, G., Kutz, S. J., Lejeune, M., St-Laurent, M. H. & Pelletier, F. Parasite prevalence, infection intensity and richness in an endangered population, the Atlantic-Gaspésie caribou. Int. J. Parasitol. Parasites Wildl. 7, 90 (2018).
Stauffer, W. & Ravdin, J. I. Entamoeba histolytica: an update. Curr. Opin. Infect. Dis. 16, 479–485 (2003).
Matsubayashi, M. et al. First detection and molecular identification of Entamoeba bovis from Japanese cattle. Parasitol. Res. 117, 339–342 (2018).
Jacob, A. S., Busby, E. J., Levy, A. D., Komm, N. & Clark, C. G. Expanding the Entamoeba universe: new hosts yield novel ribosomal lineages. J. Eukaryot. Microbiol. 63, 69–78 (2016).
Betts, E. L. et al. Genetic diversity of Blastocystis in non-primate animals. Parasitology. 145, 1228–1234 (2018).
Maloney, J. G., Jang, Y., Molokin, A., George, N. S. & Santin, M. Wide genetic diversity of Blastocystis in white-tailed deer (Odocoileus virginianus) from Maryland, USA. Microorganisms 9, (2021).
Kaczmarek, A. et al. Blastocystis occurrence and subtype diversity in wild European terrestrial mammals – the case of Białowieża Primeval Forest (NE Poland). Int. J. Parasitol. Parasites Wildl. 16, 120–125 (2021).
Wang, J. et al. Distribution and genetic diversity of Blastocystis subtypes in various mammal and bird species in northeastern China. Parasit. Vectors. 11, 522 (2018).
Kelly, R. P., Shelton, A. O. & Gallego, R. Understanding PCR processes to draw meaningful conclusions from environmental DNA studies. Sci. Rep. 9, 12133 (2019).
Padbury, G. A., Acton, D. F., Stushnoff, C. T., University of Regina. Canadian Plains Research Center, & Saskatchewan. Saskatchewan Environment and Resource Management. The Ecoregions of Saskatchewan, p. 205 (University of Regina, 1998). https://books.google.ca/books/about/The_Ecoregions_of_Saskatchewan.html?id=yRHhi6BtXfEC&redir_esc=y.
Thomas, D. C., Edmonds, E. J. & Brown, W. K. The diet of woodland caribou populations in west-central Alberta. Rangifer. 337–342. https://doi.org/10.7557/2.16.4.1275 (1996).
Gautier, C., Langlois, A., Sasseville, V., Neave, E. & Johnson, C. A. Remote sensing, snow modelling, survey data and indigenous knowledge show how snow and sea-ice conditions affect Peary caribou (Rangifer tarandus pearyi) distribution and inter-island and island–mainland movements. Polar Res. 41. https://doi.org/10.33265/polar.v41.79 (2022).
Kivinen, S., Moen, J., Berg, A. & Eriksson, Å. Effects of modern forest management on winter grazing resources for reindeer in Sweden. AMBIO. 39, 269–278 (2010).
Grenet, E., Breton, A., Barry, P. & Fonty, G. Rumen anaerobic fungi and plant substrate colonization as affected by diet composition. Anim. Feed Sci. Technol. 26, 55–70 (1989).
Gao, A. W., Wang, H. R., Yang, J. L. & Shi, C. X. The effects of elimination of fungi on microbial population and fiber degradation in sheep rumen. Appl. Mech. Mater. 295–298, 224–231 (2013).
Glendinning, L., Genç, B., Wallace, R. J. & Watson, M. Metagenomic analysis of the cow, sheep, reindeer and red deer rumen. Sci. Rep. 11, 1990 (2021).
Zielińska, S., Kidawa, D., Stempniewicz, L., Łoś, M. & Łoś, J. M. New insights into the microbiota of the Svalbard reindeer Rangifer tarandus platyrhynchus. Front. Microbiol. 7, 170. https://doi.org/10.3389/fmicb.2016.00170 (2016).
Henderson, G. et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 5, 14567 (2015).
Wu, X. et al. Characterizing the alteration in rumen microbiome and carbohydrate-active enzymes profile with forage of muskoxen rumen through comparative metatranscriptomics. Microorganisms. 10, 71 (2022).
Perea, K. et al. Feed efficiency phenotypes in lambs involve changes in ruminal, colonic, and small-intestine-located microbiota1. J. Anim. Sci. 95, 2585–2592 (2017).
Patra, A. K. & Yu, Z. Genomic insights into the distribution of peptidases and proteolytic capacity among Prevotella and Paraprevotella species. Microbiol. Spectr. 10, e02185–e02121 (2022).
Moore, S. L. & Wilson, K. Parasites as a viability cost of sexual selection in natural populations of mammals. Science. 297, 2015–2018 (2002).
Williams, A. R. et al. Emerging interactions between diet, gastrointestinal helminth infection, and the gut microbiota in livestock. BMC Vet. Res. 17, 62 (2021).
Acknowledgements
The authors thank Bridget Redquest for preparing and shipping the samples, as well as Caroline Bourdon, Julianne Mathon-Dufour, Camille Bernier, and Marie-Josée Morency for technical support in the laboratory. Funding from this research was provided by the Genomic Applications Partnership Program of Genome Canada and the Cumulative Effect program of the Canadian Forest Service.
Author information
Authors and Affiliations
Contributions
C.M. and M.M. conceived the study. M.M, P.W., C.M. and A.D. acquired funding. G.P., M.M. and P.W. provided samples and metadata. P.G. developed a data analysis tool. M.N., MJ.B. and P.G. collected data and conducted analyses under C.M. supervision. All co-authors contributed to data interpretation. M.N. wrote the manuscript. All other co-authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nagati, M., Bergeron, MJ., Gagné, P. et al. Exploring winter diet, gut microbiota and parasitism in caribou using multi-marker metabarcoding of fecal DNA. Sci Rep 14, 27960 (2024). https://doi.org/10.1038/s41598-024-76594-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-76594-5
